Multiplet States¶
Calculations with ADF yield results for one-determinant electronic states, which are not always the ‘true’ states of the molecule. The evaluation of the correct multiplet energies is not trivial in this approach, see further below the section on multiplet energies. The point is to evaluate a specific multiplet state as a linear combination of selected one-determinant functions, each computed in the field of the so-called Average-of-Configuration (AOC). Typically, in an open shell system, the AOC is the spin-restricted system in which all orbitals in the open shell are degenerate and equally occupied. The AOC serves then as a fragment for the subsequent calculations, in which the different open shell orbitals are occupied differently by specifying the appropriate occupation numbers as explained below.
Important: in these follow-up calculations it is imperative that the results are obtained in the AOC field: no SCF convergence must be carried out, because we only want to assign the electrons differently, while keeping exactly the AOC orbitals. To achieve this, the follow-up calculations must use the keyword SCF, and the subkey iterations must be set to 0.
Since ADF requires that the point-group symmetry matches not only to the nuclear frame but also to the electronic charge density and MO occupations, these calculations must run in a lower point group symmetry. Often you will also want to run the modified calculations spin-unrestricted. For an example, see the set of sample runs that come with the package and the discussion in the Examples.
The calculation of the one-determinant states based on the AOC reference state is controlled with the key SLATERDETERMINANTS, This is a general key; it can be used as a simple key and requires an argument then. It can also be used as a block key. For this particular key it is not correct to specify an argument and a data block.
SLATERDETERMINANTS file
When used as a simple key, the argument must be a file (including the path). The file must be an ASCII file containing data in the same format as you would supply in the data block when using the key as block type key, see below. All information on the file until the eof must be suitable for the data block, but no record ‘end’ on the file must be specified: only the contents of the data block.
The block format:
SLATERDETERMINANTS file
title1
irrep occups
irrep occups
...
subend
title2
irrep occups
...
subend
title3
...
subend
...
end
Each ‘title’ functions as a subkey, but is otherwise an arbitrary string to label the resulting one-determinant calculation. Each such subkey block contains the occupation numbers for a single one-determinant calculation. It is necessary that the calculation uses the reference AOC run as its only fragment file. The occupations in the subkey blocks must be re-arrangements of the AOC open-shell electrons. In the Slaterdeterminants calculation you must explicitly specify the point group symmetry in which you want to run; this must be a lower symmetry than the AOC one, otherwise you couldn’t rearrange the open shell electrons. See the sections below on multiplet energies. An sample run is included in Examples.
Each ‘irrep occups’ record specifies the occupations for the indicated irrep in the usual way (see for instance the occupations key). The irrep labels must correspond to the (lower) point group symmetry used in the slaterdeterminants calculation. Note that in an unrestricted calculations, occupations numbers must be given for both spins, using the double slash (//) to separate the occupations for spin-\(\alpha\) and spin-\(\beta\).
In this setup, the program will for each of the subkey blocks under the slaterdeterminants key execute an SCF calculation with only one cycle, i.e. no convergence, where the start-up field is the fragment field, i.e. the AOC field. So all one-determinant states in this calculation are evaluated in the AOC field. The resulting energies for the distinctly computed one-determinant states can then be combined to the desired multiplet values, corresponding to how the multiplet states are combinations of the one-determinant states.
Multiplet energies¶
The energies of atomic and molecular multiplet states that correspond to a given electron configuration can be calculated approximately with the method suggested in ref. [323]. There it is indicated that it would not be justified to take an arbitrary configuration-state function (CSF), defined in general as a linear combination of determinants that has specific spin and space symmetry properties, and use the corresponding alpha and beta spin densities in a DFT energy expression. The same holds true for the densities corresponding to the average-of-configuration (see section ‘DFT energy of a one-determinantal wave function’, the ‘procedure’ notes).
Therefore, we restrict ourselves to just computing the DFT energies of single-determinant wavefunctions. Usually (but not always) this is sufficient information to obtain the multiplet energies. The procedure, which is explained in [323], requires knowledge of the CSFs belonging to a given configuration. This means that a multiplet state with specific L, ML and S, MS values has to be written as a linear combination of the determinant wavefunctions that belong to the given configuration. With the auxiliary program ASF (Adf Single-determinants Fribourg, developed by Claude Daul in Fribourg, Switzerland) all the CSFs can be obtained, printed as linear combinations of the determinants [324]. The inverse transformation yields the determinants written as linear combinations of the CSFs.
It is often advantageous to search for CSFs that consist of one determinant only, since the energy of this determinant should correspond directly to the multiplet energy. Sometimes there is redundancy in the information and there may even be some inconsistency: two determinants may exist that both are CSFs belonging to the same multiplet state but yield somewhat different energies. We will illustrate this for the Carbon atom example treated below.
The discrepancies are a measure of ‘error bars’ associated with the theoretical multiplet energies. As a matter of fact, there are certain symmetry relations between the energies of the determinants of a configuration, calculated as the expectation value of the determinant for the full many-electron Hamiltonian. An example is the equal energy for the determinants of a \(p^1\) configuration, whether the electron is placed in the \(p_0\) (= \(p_z\)) orbital or in the \(p_{+1} (=(p_x + i p_y) / \sqrt{2})\) orbital. This equality is not obtained with present-day density functionals, leaving an ambiguity (‘error bar’) in the determination of the energy. A more complete treatment of the symmetry relations between determinant energies is given in [324].
The auxiliary program ASF, that for finite point groups finds the CSFs as linear combinations of determinants, performs also a symmetry analysis of all the two-electron integrals for a configuration, reducing them to a minimum number of non-redundant ones. ASF expresses the energies of the multiplets in the non-redundant two-electron integrals.
Warning: However, there have occasionally been found inconsistent results. A comparison to the results obtained by the procedure outlined in [323] may show significant differences and the latter seem more accurate and consistent.
DFT energy of a one-determinantal wave function
The determinant corresponds to a well defined \(\rho^\alpha\) and \(\rho^\beta\). Suppose we are dealing with a \(d^2\) configuration and we wish to know the energy of
where \(d_m\) has the \(Y_{2m}\) angular part. This determinant is a CSF of the \(^3 F\) multiplet
We can easily transform to the real spherical harmonics that are used in ADF:
with back transformations:
Here the superscripts c and s stand for the cosine, respectively sine type of combinations of \(exp(-im \phi)\) and \(exp(im \phi)\). This yields explicitly:
For \(D_1\) we obtain:
The fractional occupations have to be used in order to generate the densities ρα and ρβ and the corresponding density matrices \(\rho_\alpha\) and \(\rho_\alpha\). The density matrices can be used to calculate the energy of D1 (and \(^3 F\)) with respect to the energy of the ‘master fragment’, which should be the restricted atom with \(d^2\) occupation. Other determinants of this configuration can be treated similarly to obtain more multiplet energies of the \(d^2\) configuration.
Below is an example of how you can obtain determinant energies ‘by hand’, i.e. by carrying out a specific sequence of ADF calculations. ADF supports an automatic procedure to do this, using the input key SLATERDETERMINANTS, see the ADF User’s Guide, the Examples, and below.
Procedure
- Determine a set of orbitals belonging to the given configuration. These orbitals are generated in what we call the average-of-configuration (AOC) calculation. This is a spin-restricted SCF calculation where the electrons of the configuration are distributed equally over the subspecies of the open shell irreps in order to retain the A1 symmetry of the total density in the symmetry group of the molecule. For instance, in case of an atomic d2 configuration, the AOC calculation can be done in symmetry atom with occupation 2 in the d irrep. In case of an t2g 5 eg 1 configuration of an octahedral complex, the AOC calculation requires an occupation of 5 electrons in the t2g , and 1 electron in the eg . The result file TAPE21 of the AOC calculation has to be saved, to be used as a fragment file in the subsequent calculations.
- The AOC is used as a fragment in all subsequent calculations that are performed to obtain single determinant energies. This means that those single determinant energies are always with respect to the AOC energy. This is a case where there is only one “fragment”, which is actually the complete system, but in a different electronic configuration and in possibly a different symmetry group.
Suppose that a single determinant corresponds to spin-up and spin-down densities \(\rho^\alpha\) and \(\rho^\beta\) , i.e. to specific spin-unrestricted occupations of the AOC orbitals. These densities \(\rho^\alpha\) and \(\rho^\beta\) correspond to a symmetry group that will in general be a subgroup of the symmetry group of the molecule. For instance, the occupation (p+1 \(\alpha\))1 in the case of an atomic p1 configuration corresponds to
with D∞h symmetry.
To obtain the energy of the determinant wave function we must now perform one cycle (iterations= 0 in the block key SCF) of a spin-unrestricted calculation, with AOC as (the only) fragment with alpha and beta occupation numbers (using the input key occupations) such that \(\rho^\alpha\) and \(\rho^\beta\) result. Note that the appropriate (lower) symmetry point group must be specified in the input file.
Occasionally, the single determinant corresponds to a closed shell configuration in the appropriate lower symmetry, for instance determinant D10 = |0+ 0- | of the p2 configuration of Carbon, with density r=pz 2 . In that case the one-cycle calculation can of course be spin-restricted.
N.B.1. One cycle will regenerate the SCF orbitals of AOC, if the same field is used as the converged AOC field. This will actually be the case because the starting potential is taken from the fragment TAPE21 file. The key modifystartpotential must not be used (the density should be distributed equally over the spins).
N.B.2. After diagonalization in the one-cycle run, the AOC orbitals have been obtained again and are occupied as specified. The (‘bonding’) energy is calculated from the resulting charge density.
Remarks:
- If one does not perform just one cycle, but instead converges the unrestricted calculation, the energy will be lowered by ‘polarization’ of the orbitals. It is theoretically not so clear what the status of such converged energies is. Usually the energy lowering is in the order of 0.1 eV, but it may be quite a bit larger.
- It is not necessary to use AOC as fragment in the single-determinant runs. It is also perfectly allowed to run all calculations (ground state, AOC, determinants) from one set of fragments, for instance the standard atomic fragments. Since we must arrange that the one-cycle determinant calculations use the AOC field, so as to reproduce the AOC orbitals, we must then supply the result file TAPE21 of the AOC run as a restart file, using the key restart; see the adf User’s Guide. Of course, in such an approach the computed energies are with respect to another reference, for instance the restricted atoms.
Results for first period atoms
In one of the next sections tables are given for the energy lowering in going from the converged spherically symmetric spin-restricted atom (the ‘master’ fragment) to specific one-determinant wavefunctions with the orbital occupations as specified. Note that the px and py populations are always equal; only their sum is given. In many cases the determinant corresponds to a specific state, which is then given in the last column. For each atom, the first calculation is for the spherically symmetric spin-unrestricted atom. These tables are now obsolete, all information needed to obtain the atomic reference energies, i.e. the ground state multiplet energy with respect to the AOC, can be found in ref. [325].
Examples worked out for all first period atoms:
H: Configuration (1s)1. Only one determinant: | 1s \(\alpha\) (1) |
He: Configuration (1s)2 . Closed shell.
Li: Configuration (2s)1 . Only one determinant: | 2s \(\alpha\) (1) |
Be: Configuration (2s)2 . Closed shell.
- B: Configuration (2p)1 .
Ground multiplet 2 P.
\[\begin{split}D_1 & = |p_1 \alpha (1) | = | ^2 P ; M_L =1 ; M_S = 1/2 \rangle \\ \rho^\alpha & = 1/2(p_x-ip_y)(p_x+ip_y) = 1/2(p_x^2+p_y^2)\end{split}\]The occupation numbers for D1 are
\[\begin{split}p_x^\alpha & = p_y^\alpha = 1/2 \\ p_z^\alpha & = p_x^\beta = p_y^\beta = p_z^\beta = 0\end{split}\]Another determinant belonging to 2 P is D2 = |..p0 \(\alpha\) (1)| with occupations \(p_z^\alpha = 1\) and all other p-occupations zero. This determinant is 0.04 eV lower in energy than D1 for LDA, but 0.15 eV for BP.
- C: Configuration p2 .
Multiplet states are 3 P, 1 S and 1 D. We use this atom as an example of the general procedure. First write down all determinants belonging to p2 and group them according to MS and ML (1+ \(\equiv\) p1 \(\alpha\), ...)
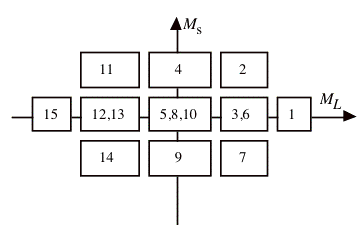
Determinant MS ML D1 = |1+ 1- | 0 2 D2 = |1+ 0+ | 1 1 D3 = |1+ 0- | 0 1 D4 = |1+ -1+ | 1 0 D5 = |1+ -1- | 0 0 D6 = |1- 0+ | 0 1 D7 = |1- 0- | -1 1 D8 = |1- -1+ | 0 0 D9 = |1- -1- | -1 0 D10 = |0+ 0- | 0 0 D11 = |0+ -1+ | 1 -1 D12 = |0+ -1- | 0 -1 D13 = |0- -1+ | 0 -1 D14 = |0- -1- | -1 -1 D15 = |-1+ -1- | 0 -2 
The presence of a determinant with ML = 2, MS = 0 and no ML = 2, MS 0 determinant indicates the presence of a 1 D multiplet, and E(1 D) = E(D1 ). There is also a 3 P, the determinant with MS = 1, ML = 1. We should have E(3 P) = E(D2 ) = E(D4 ). The two determinants D3 and D6 in the MS = 0, ML = 1 box will mix, and the solutions of the 2 by 2 secular problem will be E(1 D) and E(3 P). Since the sum of the eigenvalues is equal to the sum of the initial diagonal elements, we have E(1 D) + E(3 P) = E(D3 ) + E(D6 ). We should also have E(D3 ) + E(D6 ) = E(D1 ) + E(D2 ). Such a relation provides a test on the consistency of the results. Finally we have the MS = 0, ML = 0 block. The sum of the energies of D5 , D8 and D10 should be the sum of the energies of 1 S, 3 P and 1 D. Since E(3 P) and E(1 D) are already known, E(1 S) can be calculated.
In the appendix we first locate for C the spherical unrestricted atom. Next we have E(D4 ), yielding E(3 P) = -1.345 eV (LDA + Becke). Next E(D2 ) = E(3 P) = -1.189 (always LDA + Becke). The difference is substantial: ~ 0.15 Next we have E(D3 ) = - 0.812. Since E(D6 ) = E(D3 ), because \(\rho^\alpha\) (D6 ) = \(\rho^\beta\) (D3 ) and \(\rho^\beta\) (D6 ) = \(\rho^\alpha\) (D3 ), we should have 2E(D3 ) = -1.624 = E(1 D) + E(3 P). Therefore E(1 D) = -1.624 - (-1.345) = - 0.279 or E(1 D) = - 1.624 - (-1.189) = - 0.435. These numbers can be checked against E(D1 ) which also should be E(1 D): E(D1 ) =+ 0.044. The discrepancy is large! Finally, 1 S can be obtained: E(D10 ) = + 0.319 (D8 ) = E(D1 ) = + 0.044 E(D5 ) = E(D1 ) = + 0.044. So 0.407 = E(1 S) + E(3 P) +E(1 D). Different results for E(1 S) are obtained depending on the E(3 P) and E(1 D) we choose: e.g. E(1 S) = 0.407 -(-1.345) - (- 0.279) = 2.031 or E(1 S) = 0.407 - (-1.189) - (0.044) = 1.552. Comparing to experiment we might calculate the excitation energies w.r.t. E(3 P):
calculated experimental HF 3 P → 1 D: 1.066 to 1.389 1.26 1.55 3 P → 1 S: 2.741 to 3.376 2.684 3.78 - N: Configuration p3 .
- Ground multiplet 4 S, corresponds to the spherical unrestricted atom, energy -2.943 eV. Other multiplets: 2 P, 2 D. According to the printed output for configuration p3 we have |2 D;ML =2;MS =1/2 = |p1 \(^\alpha\) p1 \(^\beta\) p0 \(^\alpha\) | = D2 \(\rho^\alpha\) = 1/2 px 2 + 1/2 py 2 + pz 2 \(\rho^\beta\) = 1/2 px 2 + 1/2 py 2 E(D2 ) = - 0.745 according to the table in the Appendix (LDA + Becke). The energy of D1 , with \(\rho^\alpha\) = px 2 + py 2 , \(\rho^\beta\) = pz 2 , is E(|1A 2B 3A|) = -1.9702. The energy of D3 , with \(\rho^\alpha\) = 1/2 px 2 + 1/2 py 2 + pz 2 , \(\rho^\beta\) = pz 2 corresponding to |1A 2A 2B| or |2A 2B 3A|, is E(D3 ) = - 0.158. Finally, D4 has \(\rho^\alpha\) = px 2 + py 2 , \(\rho^\beta\) = 1/2 px 2 + 1/2 py 2 , corresponding to |1A 1B 3A| and |1A 3A 3B|, and E(D4 ) = - 0.109. The ML = 1, MS =1/2 determinants are |1A 1B 3A| and |1A 2A 2B|. Therefore E(2 D) + E(2 P) = E(D4 ) + E(D3 ), so E(2 P) = - 0.109 - 0.158 - (- 0.745) = + 0.478. We can use D1 in the ML =0, MS =1/2 block, from which we find E(4 S) + E(2 D) + E(2 P) = 2E(D2 ) =+ E(D1 ). Hence E(2 P) = -1.490 - 1.9702 - (- 0.745) - (- 2.943) = + 0.2278.
- O: Configuration p4 .
- Multiplet states 3 P, 1 S, 1 D. D1 , with \(\rho^\alpha\) = px 2 + py 2 + pz 2 , \(\rho^\beta\) = pz 2 corresponds to |1A 2A 2B 3A| , the ML =1, MS =1 determinant of 3 P: E(3 P) = -1.836 D2 with \(\rho^\alpha\) = px 2 + py 2 + pz 2 , \(\rho^\beta\) = 1/2 px 2 + 1/2 py 2 , corresponds to |1A 1B 2A 3A|, the determinant of 3 P: E(3 P) = -1.568 D3 , with \(\rho^\alpha\) = 1/2 px 2 + 1/2 py 2 + pz 2 , \(\rho^\beta\) = px 2 + py 2 , corresponds to |1A 1B 2A 3B|, and ML =1, MS =0 belonging to 3 P as well as 1 D.
- F: Configuration p5 .
- Ground multiplet 2 P. As in B, we have two determinants with different energies belonging to this state: D1 = |...(p0 \(^\alpha\) )1 (p0 \(^\beta\) )0 | → E(D1 ) = - 0.715. D2 = |(p-1 )2 (p0 )2 (p1 \(^\alpha\) )1 (p1 \(^\beta\) )0 | → E(D2 ) = - 0.467.
Ne: Configuration p6 . Closed shell.
Ground and Excited State Multiplets
The computation of multiplets can be carried out with adf, using the input key SLATERDETERMINANTS.
The method described in [323] for the calculation of the energies of proper spin and spatial symmetry adapted Configuration State Functions is based on the calculation of the energies of single determinantal wavefunctions. The densities corresponding to those Slater determinants are inserted in the approximation used for the exchange-correlation energy.
The procedure as detailed above is somewhat involved. Moreover, one would like to have an easy procedure to calculate many determinants. This is particularly desirable if one wishes to calculate the energies of all Slater determinants of a given configuration, for instance if one wishes to calculate certain averages in view of the inconsistencies of the method.
We have implemented a semi-automatic procedure, using the key SLATERDETERMINANTS.
The general idea of this method is to first perform a restricted calculation in the symmetry that is appropriate for the molecule. This is called the average-of-configuration (AOC) calculation. This AOC calculation generates the orbitals which will be used in all the Slater determinants.
The AOC is the fragment that must be used in all subsequent calculations. The subsequent calculations are characterized by having the AOC as the (only) fragment, and by specifying the keyword SLATERDETERMINANTS. This key is a general key: it may be used as a simple key (it must then have an argument) or as a block type key (no argument, but a data block). If an argument is given this must be a file name. The named file should contain the occupations for the determinants (see below). If no file name is specified the key should be a block key and the occupations should be specified in the data block.
The required information, on file or in the data block, is the specification of the determinant or determinants that are to be calculated in the form of orbital occupation numbers for the AOC orbitals, using the irrep labels of the point group of the AOC calculation, see below for format. All specified determinants will be calculated, and the obtained energy will always be the energy difference with respect to the AOC. Default occupations for all subspecies of the AOC fragment are the occupations of the AOC itself. Therefore, only the open (modified) subspecies have to be specified.
One has to be careful with respect to the point group symmetry to use in the SLATERDETERMINANTS calculation. The density belonging to a specific determinant is usually lower than the AOC symmetry (which is the full symmetry group of the system). In that case this lower point group symmetry has to be specified in the SLATERDETERMINANTS calculation. Everything will always work fine if one just does not use any symmetry at all (nosym). However, if for reasons of computational efficiency one does want to use a subgroup of the system that corresponds to the determinant density or densities, this is perfectly possible. However, all the densities of the determinants specified must then have this (or a higher) symmetry. The program does not check on this, it is the user’s responsibility to make sure that this condition is satisfied for all the determinants. The only check that is performed is that occupations of equivalent representations (subspecies of one irrep) in the lower point group of the SLATERDETERMINANTS run, that result from the specified occupations of the subspecies of the AOC symmetry, are equal.
Format of the input.
Important: in the SlaterDeterminants calculations it is imperative that the results are obtained in the AOC field: no SCF convergence must be carried out, because we only want to assign the electrons differently, while keeping the AOC orbitals exactly as they are. To achieve this, the calculations should use the keyword SCF, and the subkey iterations has to be set to 0 in the SCF data block.
Since adf requires that the point-group symmetry conforms not only to the nuclear frame but also to the electronic charge density and mo occupations, these calculations must run in a lower point group symmetry. Often you will also want to run the modified calculations spin-unrestricted.
For an example, see the set of sample runs that come with the package and the comments in the Examples document.
The calculation of the one-determinant states based on the AOC reference state is controlled with the key SLATERDETERMINANTS. It is a general key; it can be used as a simple key and requires an argument then. It can also be used as a block key. For this particular key it is not correct to specify an argument and a data block.
SLATERDETERMINANTS file
When used as a simple key, the argument must be a file (including the path). The file must be an ascii file containing data in the same format as you would supply in the data block when using the key as block type key, see below. All information on the file until the eof must be suitable for the data block, but no record ‘end’ on the file must be specified: only the contents of the data block.
The block format:
SLATERDETERMINANTS
TITLE1
irrep occups
irrep occups
SUBEND
TITLE2
irrep occups
SUBEND
TITLE3
irrep occups
SUBEND
END
Each ‘title’ functions as a subkey, but is otherwise an arbitrary string to label the resulting one-determinant calculation. Each such subkey block contains the occupation numbers for a single one-determinant calculation. It is necessary that the calculation uses the reference AOC run as its only fragment file. The occupations in the subkey blocks must be re-arrangements of the AOC open-shell electrons. In the SLATERDETERMINANTS calculation you must explicitly specify the point group symmetry in which you want to run. The \(\rho^\alpha\) and \(\rho^\beta\) densities of all determinants to be calculated must have this point group symmetry, or a higher symmetry.
Each ‘irrep occups’ record specifies the occupations for the indicated irrep in the usual way (see for instance the occupations key). The irrep labels must correspond to the AOC point group symmetry used in the AOC calculation, not the point group symmetry used in the ``SLATERDETERMINANTS`` calculation!. Note that in an unrestricted calculations, occupations numbers must be given for both spins, using the double slash (//) to separate the occupations for spin-alpha and spin-beta.
In this setup, the program will for each of the subkey blocks under the SLATERDETERMINANTS key execute an SCF calculation with only one cycle, i.e. no convergence, where the start-up field is the fragment field, i.e. the AOC field. So all one-determinant states in this calculation are evaluated in the AOC field. The resulting energies for the distinctly computed one-determinant states can then be combined to the desired multiplet values.
Example: Carbon \(p^2\)
SlaterDeterminants
C(p2) ALFA: s=1, px=py=2/3, pz=2/3; BETA: s=1, p=0 ! title
S 1 // 1 ! irrep name and occupations
P:x 0.666666666666666666 // 0 ! another irrep, et cetera
P:y 0.666666666666666666 // 0
P:z 0.666666666666666666 // 0
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
C(p2) ALFA: S=1, px=py=1, pz=0; BETA: s=1 !next (Sl.Determinant) title
S 1 // 1
P:x 1 // 0
P:y 1 // 0
P:z 0 // 0
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
C(p2) ALFA: s=1, px=py=0.5, pz=1; BETA: s=1
S 1 // 1
P:x 0.5 // 0
P:y 0.5 // 0
P:z 1 // 0
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
C(p2) ALFA: s=1, px=py=0.5, pz=0; BETA: s=1, px=py=0, pz=1
S 1 // 1
P:x 0.5 // 0
P:y 0.5 // 0
P:z 0 // 1
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
C(p2) ALFA: s=1, px=py=0.5, pz=0; BETA: s=1, px=py=0.5, pz=0
S 1 // 1
P:x 0.5 // 0.5
P:y 0.5 // 0.5
P:z 0 // 0
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
C(p2) ALFA: s= 1, px=py=0, pz=1; BETA: s=1, px=py=0, pz=1
S 1 // 1
P:x 0 // 0
P:y 0 // 0
P:z 1 // 1
D:z2 0 // 0
D:x2-y2 0 // 0
D:xy 0 // 0
D:xz 0 // 0
D:yz 0 // 0
SUBEND
In the example the AOC calculation is the Carbon atom in spherical symmetry (symmetry name atom).
Several spin states can be generated from this AOC set of orbitals, but they all have a lower symmetry than the AOC. In the example the point group D∞h (DLIN) could be used in the SLATERDETERMINANTS calculation. In D∞h the p orbitals split into two sets, p*x * and p*y * occur in \(\pi\)x and \(\pi\)y respectively, so their occupations must be identical, and p*z * is a \(\Sigma\)u orbital.
In the data block of the SLATERDETERMINANTS key (or in the file) we now specify the occupations for the subspecies of the atom irreps of a specific Slater determinant and the program will sort out the corresponding occupations in the d(lin) symmetry.
In all cases the orbitals used for the energy calculation(s) will be the self-consistent AOC orbitals.
In the given example, the first set of occupations does not correspond to a Slater determinant, but is the spin-polarized spherical case with the p electrons evenly distributed over all components.
LDA results, with and without GGA (Becke-Perdew)
Energy changes (eV) for atoms going from restricted to (one-cycle) unrestricted. Results between parentheses are for converged unrestricted calculations)
All calculations have been performed in D∞h symmetry, since px and py always had equal occupations and therefore could occur as \(\pi\)u -x and \(\pi\)u -y partners of the \(\Pi\)u irrep.
| El. | Occupations | LDA | LDA+Becke | BP | |||||
| alpha-spin | beta-spin | ||||||||
| s | px+py | pz | s | px+py | pz | ||||
| H | 1 | 0 | 0 | 0 | 0 | 0 | -0.868 (-0.898) | -0.758 (-0.837) | -0.889 (-0.948) |
| Li | 1 | 0 | 0 | 0 | 0 | 0 | -0.231 (-0.235) | -0.195 (-0.207) | -0.249 (-0.256) |
| Be | 1 | 0 | 0 | 1 | 0 | 0 | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) |
| B | 1 | 2/3 | 1/3 | 1 | 0 | 0 | -0.247 (-0.255) | -0.231 (-0.242) | -0.276 (-0.281) |
| 1 | 0 | 1 | 1 | 0 | 0 | -0.295 (-0.321) | -0.436 (-0.474) | -0.448 (-0.485) | |
| 1 | 1 | 0 | 1 | 0 | 0 | -0.266 (-0.279) | -0.296 (-0.316) | -0.333 (-0.348) | |
| C | 1 | 4/3 | 2/3 | 1 | 0 | 0 | -1.163 (-1.203) | -1.109 (-1.158) | -1.252 (-1.285) |
| 1 | 2 | 0 | 1 | 0 | 0 | -1.152 (-1.211) | -1.271 (-1.345) | -1.372 (-1.436) | |
| 1 | 1 | 1 | 1 | 0 | 0 | -1.152 (-1.197) | -1.134 (-1.189) | -1.267 (-1.307) | |
| 1 | 1 | 0 | 1 | 0 | 1 | -0.462 (-0.506) | -0.726 (-0.812) | -0.778 (-0.868) | |
| 1 | 1 | 0 | 1 | 1 | 0 | 0.159 (0.150) | 0.039 (0.044) | 0.087 (0.086) | |
| 1 | 0 | 1 | 1 | 0 | 1 | 0.730 (0.668) | 0.322 (0.319) | 0.480 (0.450) | |
| N | 1 | 2 | 1 | 1 | 0 | 0 | -2.936 (-3.032) | -2.827 (-2.943) | -3.101 (-3.190) |
| 1 | 2 | 0 | 1 | 0 | 1 | -1.362 (-1.454) | -1.811 (-1.972) | -1.943 (-2.108) | |
| 1 | 1 | 1 | 1 | 1 | 0 | -0.581 (-0.618) | -0.688 (-0.745) | -0.746 (-0.801) | |
| 1 | 1 | 1 | 1 | 0 | 1 | 0.178 (0.088) | -0.104 (-0.158) | -0.069 (-0.140) | |
| 1 | 2 | 0 | 1 | 1 | 0 | 0.197 (0.135) | -0.077 (-0.109) | -0.011 (-0.053) | |
| O | 1 | 2 | 1 | 1 | 2/3 | 1/3 | -1.400 (-1.477) | -1.361 (-1.447) | -1.480 (-1.552) |
| 1 | 2 | 1 | 1 | 0 | 1 | -1.442 (-1.583) | -1.698 (-1.836) | -1.816 (-1.957) | |
| 1 | 2 | 1 | 1 | 1 | 0 | -1.422 (-1.515) | -1.470 (-1.568) | -1.590 (-1.678) | |
| 1 | 1 | 1 | 1 | 2 | 0 | -0.564 (-0.623) | -0.866 (-0.960) | -0.913 (-1.013) | |
| 1 | 1 | 1 | 1 | 1 | 1 | 0.358 (0.321) | 0.255 (0.237) | 0.292 (0.266) | |
| 1 | 2 | 0 | 1 | 2 | 0 | 1.323 (1.220) | 0.825 (0.789) | 0.992 (0.932) | |
| F | 1 | 2 | 1 | 1 | 4/3 | 2/3 | -0.374 (-0.398) | -0.366 (-0.391) | -0.394 (-0.416) |
| 1 | 2 | 1 | 1 | 2 | 0 | -0.323 (-0.409) | -0.605 (-0.686) | -0.627 (-0.715) | |
| 1 | 2 | 1 | 1 | 1 | 1 | -0.349 (-0.389) | -0.401 (-0.441) | -0.427 (-0.467) | |