Setting up a QM/MM Simulation: a ‘Walk Thru’¶
In this section we provide a detailed ‘walk thru’ of the process of setting up an ADF QM/MM simulation. There will be two examples, the first being a fairly straightforward example and the second one being fairly complex.
Example A: Cytocine¶
This is a straightforward example, where the input necessary to perform a QM/MM simulation of cytosine (Figure 5-1) will be constructed.
Step 1. Partitioning the System and the Model QM system¶
First one must decide where to partition the system into QM and MM regions. This is actually a very important step since the partitioning can be considered the ‘original sin’. Much thought and testing should be put into deciding where to place the QM/MM boundary. In this example, we have chosen the partitioning depicted in Figure 5-1a in order to keep the example simple. In this figure the QM region enclosed in the dotted polygon, with two covalent bonds crossing the QM/MM boundary. One must also choose an appropriate QM model system for which the electronic structure calculation will be performed. To preserve the sp3 hybridization of the carbon center in the QM region, we must keep the carbon tetravalent. Thus, we will cap the two dangling bonds with dummy or capping hydrogen atoms. One can use any monovalent atom such as H or F, but H is probably best. The reason that monovalent atoms should be used for capping atoms is that one does not want capping atom to have any ‘dangling’ bonds. Capping or dummy groups can not be used. Figure 5-1b, depicts the QM model system with two capping hydrogen atoms. Thus, the electronic structure calculation will be performed on methane such that the capping hydrogen atoms lie along the bond vector of the link bond in the real system as shown in Figure 5-1b.
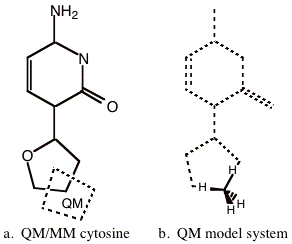
Figure 5-1 Cytocine QM/MM example model. a) Shows the whole system with the atoms enclosed in the dotted polygon making up the QM system. b) Shows the equivalent QM model system. The remainder of the cytocine molecule is shown ghosted to demonstrate the relationship between the model system and the full system. The QM model system consists of a closed shell methane molecule.
Step 2. Labeling of Atoms (QM, MM or LI)¶
Once a partitioning of the system has been established, one needs to designate each of the atoms in the full system as QM, LI or MM atom type. For the example system, these designations are shown in Figure 5-2a, where the atoms that are not labeled are MM atoms.
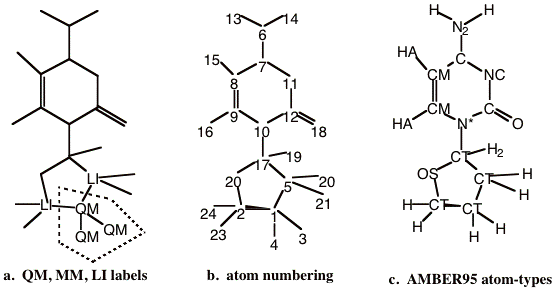
Figure 5-2 Labeling of the example model. a) QM, MM and LI designations. Atoms not labeled are ‘MM’ atoms. The dotted polygon encloses the QM region of the model. b) Atom numbering of the entire system. Note that the QM and LI atoms precede any MM atoms. c) The AMBER95 force field atom type designations.
All atoms within the dotted polygon are ‘QM’ type atoms. The atoms outside of the QM region will either be MM or LI atoms depending on whether they are part of a link bond or not. The covalent bonds that cross the QM/MM boundary are termed the link bonds. In the example system, there are two such covalent bonds. The atoms that lie on the MM side of the link bonds are labeled the LI atoms. Thus, these are all of the atoms that lie outside the dotted polygon that have a covalent bond to QM atoms. If there are no covalent bonds that traverse the QM/MM boundary, then there will be no LI atoms.
Step 3. Renumbering of Atoms¶
There is a strict rule concerning the ordering of the atoms, based on the QM, MM, or LI atom type designation. All QM and LI atoms must come before any MM atoms. A valid atom numbering for the example system is shown in figure 5-2b. Here the atoms labeled ‘QM’ or ‘LI’ in Figure 5-2a are the first five atoms of the molecular system.
Step 4. ADF QM/MM input: Atomic coordinates¶
Now we can begin to construct the input. We will begin with the atomic coordinates. For this example, we will optimize the geometry of the complex in Cartesian coordinates. Coordinates of the whole QM/MM complex or the ‘real’ complex should be defined here. DO NOT define the coordinates of the capping atoms. The program will calculate their positions, and add them automatically. The definition of the coordinates is done exactly as they are in a standard ADF run. Below is the ATOMS key block for our example system.
ATOMS Cartesian
1 C 1.94807 3.58290 -0.58162
2 C 1.94191 3.61595 1.09448
3 H 1.69949 4.49893 -1.05273
4 H 2.99455 3.17964 -0.86304
5 C 0.94659 2.40054 -0.92364
6 N -1.74397 -3.46417 0.31178
7 C -1.00720 -2.20758 0.33536
8 C -1.66928 -1.00652 0.31001
9 C -0.92847 0.25653 0.34895
10 N 0.43971 0.26735 0.38232
11 N 0.36409 -2.20477 0.28992
12 C 1.09714 -0.95413 0.22469
13 H -2.89781 -3.50815 0.31746
14 H -1.21484 -4.49217 0.31721
15 H -2.80940 -0.93497 0.30550
16 H -1.55324 1.21497 0.33885
17 C 1.23309 1.44017 0.30994
18 O 2.58277 -1.01636 0.23914
19 H 2.37276 1.25557 0.29984
20 O 1.02358 2.43085 1.50880
21 H 1.17136 1.95097 -1.87367
22 H -0.10600 2.77333 -0.80348
23 H 1.62170 4.54039 1.51392
24 H 2.99608 3.28749 1.41345
END
Step 5. Connection Table and MM force field types¶
In order to construct a molecular mechanics potential, the program needs to know the connectivity of the molecular system and the molecular mechanics force field atom-type designations. In this example we are using the AMBER95 force field of Kollman and coworkers [4]. The appropriate AMBER95 atom-types for this molecule are shown in Figure 5-2c. No new atom types need to be introduced to the standard AMBER95 force field to treat this system. However, if this were needed, then the force field file would have to be modified.
Next a connection table needs to be constructed. For this program this needs to be done on an atom by atom basis. Either a fully redundant connection table or a fully non-redundant connection table is acceptable. A redundant connection table refers to one in which the covalent bonds are defined for all atoms. For example, if X is bonded to Y, in the connections for atom X, a bond is defined to atom Y. For the connections to atom Y, a bond is also defined to atom X even though the bond has already been defined. In a non-redundant connection table, when a bond is defined in the connections for atom X, it is not again defined in the connections for atom Y.
We now can begin to construct part of the input, namely the MM_CONNECTION_TABLE subkey block of the QMMM key block. For this example, the MM_CONNECTION_TABLE key block is given below.
MM_CONNECTION_TABLE
1 CT QM 2 3 4 5
2 CT LI 1 20 23 24
3 HC QM 1
4 HC QM 1
5 CT LI 1 17 21 22
6 N2 MM 7 13 14
7 CA MM 6 8 11
8 CM MM 7 9 15
9 CM MM 8 10 16
10 N* MM 9 12 17
11 NC MM 7 12
12 C MM 10 11 18
13 H MM 6
14 H MM 6
15 HA MM 8
16 H4 MM 9
17 CT MM 5 10 19 20
18 O MM 12
19 H2 MM 17
20 OS MM 2 17
21 HC MM 5
22 HC MM 5
23 H1 MM 2
24 H1 MM 2
SUBEND
The first column is simply the atom number. The atoms defined here MUST be in the same order as defined in the ATOMS key block provided in the previous section. Again, we do not include the capping atoms. The second column shows the AMBER95 atom-types for our system, displayed in Figure 5-2c. The third column is the MM, QM or LI designation. Notice that the QM and LI atoms appear before any MM atoms. The remaining columns are reserved for the connection table. In the above example, a fully redundant connection table is provided.
Step 6. LINK_BONDS¶
When there are covalent bonds that cross the QM/MM boundary, the LINK_BONDS subkey block is required. Since one only defines the ‘real system in both the ATOMS key block and the MM_CONNECTION_TABLE subkey block, this key block defines both the initial position of the capping atom and what kind of ADF fragment atom will be used as a capping atom. In this example we have two link bonds, both of which will be ‘capped’ with capping hydrogen atoms as shown in Figure 5-1b. Below is the LINK_BONDS subkey block for our example.
LINK_BONDS
1 - 5 1.380 H
1 - 2 1.375 H
SUBEND
The first part of the input specifies the atoms involved in link bonds. Here QM atom 1 forms link bonds with atoms 5 and 2. The column in the input is the link bond a parameter, which is defined as the ratio between the capping bond length in the QM model system and the bond length of the corresponding link bond in the real system. This ratio can be determined by taking the necessary bond lengths from a pure QM calculation of the model QM system, and the bond length from the whole complex. If those are not available, they can be taken from tabulated bond lengths or bond lengths of similar bonds in other complexes. There is an independent a parameter for each link bond. It is VERY IMPORTANT to emphasize that the total energy of the QM/M system is dependent upon the a parameters. Thus, if one is comparing the energetics of two conformational isomers calculated with the QM/MM method, this comparison is only valid if the a parameters used are the same. In our example, ratios of 1.38 and 1.375 were used. This is somewhat typical ratio of C-H to C-C bond lengths, in aliphatic hydrocarbons. The last column in the LINK_BONDS input refers to the ADF fragment for which will be used for the capping atom in the electronic structure calculation. Please, note this fragment must be present in the FRAGMENTS key block of the ADF input.
Step 7. Assignment of Atomic Charges¶
Perhaps the most dubious aspect of the QM/MM approach involves the non-bonded electrostatic interaction between the QM and MM regions. The ADF QM/MM extension currently only supports placement of static point charges on MM atoms. At the moment, you have two options. First, you can chose to have the MM point charges to interact with the electron density of the QM model system, thereby allowing the wave function of the QM system to be polarized. Alternatively, you can assign static point charges to the QM atoms which interact with MM point charges as would happen if the whole system were treated with a molecular mechanics force field. In this example, we will choose the latter, using the standard AMBER95 charges cytosine. To specific how the electrostatic interactions between the two regions are treated, one uses the ELSTAT_COUPLING_MODEL keyword in the QMMM key block and sets it equal to 1.
In ADF QM/MM the atomic point charges can be assigned on an atom-type basis, where the point charges are taken from the force field file. It can also be defined on a per atom basis, where a unique charge is assigned to each atom in the molecular system in the CHARGES subkey block. Since the charges in AMBER95 are assigned according to the nucleic or amino acid, we must assign the charges on a per-atom basis. Given below is the CHARGES subkey block with the appropriate AMBER95 point charges assigned to the system. The first column in this subkey block is the atom numbering. It is important to use the right atom number instead because the program actually determines the charges on each atom individually by searching for the atom number within this key block. Charges don’t have to be in order.
ELSTAT_COUPLING_MODEL=1
CHARGES
1 0.0000
2 0.0000
3 0.0000
4 0.0000
5 0.0000
6 -0.9530
7 0.8185
8 -0.5215
9 0.0053
10 -0.0484
11 -0.7584
12 0.7538
13 0.4234
14 0.4234
15 0.1928
16 0.1958
17 0.0066
18 -0.6252
19 0.2902
20 -0.2033
21 0.0000
22 0.0000
23 0.0000
24 0.0000
SUBEND
Step 8. Remainder of the QMMM key block¶
The ADF QM/MM input is almost complete. Now only a few settings need to be defined in the QMMM key block. The remainder of the QMMM key block is given below.
FORCEFIELD_FILE /usr/bob/QMMM_data/amber95.ff
RESTART_FILE mm.restart
OUTPUT_LEVEL=1
WARNING_LEVEL=2
The FORCEFIELD_FILE defines the filename of the force field file to be used. If the force field file is not in the running directory of the ADF job, then the full path needs to be specified. The RESTART_FILE specifies the name of the QM/MM restart to be written. If the job is a restart itself, this keyword also specifies the QM/MM restart file to read.
The OUTPUT_LEVEL specifies how much output to print during the course of the ADF QM/MM run. OUTPUT_LEVEL=1 is good for most purposes. Using an OUTPUT_LEVEL=2 is good when troubleshooting, but probably provides too much output when the job is running normally. The WARNING_LEVEL keyword specifies when to stop the job. When it is set to 2, the run stops at any spot where a potential QM/MM problem is detected. This is good when first setting up a job because the program attempts to point out potential problems.
Step 9. Putting it all together: The whole ADF QM/MM input¶
The whole ADF QM/MM input for the sample system is given below. The following will be a QM/MM geometry optimization performed in Cartesian coordinates with no constraints. Some comments are provided in bold.
Title CYT amber95 test - CARTESIAN GEOMETRY OPTIMIZATION NO CONSTRAINTS
Fragments
C T21.C.III.1s ! Notice that only fragments for the calculation of
H T21.H.III ! model system are needed.
End
Symmetry NOSYM
Charge 0 0 ! This refers to the charge of the QM model system, not the 'real' system
ATOMS Cartesian
1 C 1.94807 3.58290 -0.58162
2 C 1.94191 3.61595 1.09448
3 H 1.69949 4.49893 -1.05273
4 H 2.99455 3.17964 -0.86304
5 C 0.94659 2.40054 -0.92364
6 N -1.74397 -3.46417 0.31178
7 C -1.00720 -2.20758 0.33536
8 C -1.66928 -1.00652 0.31001
9 C -0.92847 0.25653 0.34895
10 N 0.43971 0.26735 0.38232
11 N 0.36409 -2.20477 0.28992
12 C 1.09714 -0.95413 0.22469
13 H -2.89781 -3.50815 0.31746
14 H -1.21484 -4.49217 0.31721
15 H -2.80940 -0.93497 0.30550
16 H -1.55324 1.21497 0.33885
17 C 1.23309 1.44017 0.30994
18 O 2.58277 -1.01636 0.23914
19 H 2.37276 1.25557 0.29984
20 O 1.02358 2.43085 1.50880
21 H 1.17136 1.95097 -1.87367
22 H -0.10600 2.77333 -0.80348
23 H 1.62170 4.54039 1.51392
24 H 2.99608 3.28749 1.41345
END
QMMM
FORCEFIELD_FILE amber95.ff
RESTART_FILE mm.restart
OUTPUT_LEVEL=1
WARNING_LEVEL=2
ELSTAT_COUPLING_MODEL=1
LINK_BONDS
1 - 5 1.38000 H
1 - 2 1.38030 H
SUBEND
MM_CONNECTION_TABLE
1 CT QM 2 3 4 5
2 CT LI 1 20 23 24
3 HC QM 1
4 HC QM 1
5 CT LI 1 17 21 22
6 N2 MM 7 13 14
7 CA MM 6 8 11
8 CM MM 7 9 15
9 CM MM 8 10 16
10 N* MM 9 12 17
11 NC MM 7 12
12 C MM 10 11 18
13 H MM 6
14 H MM 6
15 HA MM 8
16 H4 MM 9
17 CT MM 5 10 19 20
18 O MM 12
19 H2 MM 17
20 OS MM 2 17
21 HC MM 5
22 HC MM 5
23 H1 MM 2
24 H1 MM 2
SUBEND
CHARGES
1 0.0 CT
2 0.0 CT
3 0.0 HC
4 0.0 HC
5 0.0 CT
6 -0.9530 N2
7 0.8185 CA
8 -0.5215 CM
9 0.0053 CM
10 -0.0484 N*
11 -0.7584 NC
12 0.7538 C
13 0.4234 H
14 0.4234 H
15 0.1928 HA
16 0.1958 H4
17 0.0066 CT
18 -0.6252 O
19 0.2902 H2
20 -0.2033 OS
21 0.0000 HC
22 0.0000 HC
23 0.0000 H1
24 0.0000 H1
SUBEND
END
GEOMETRY
ITERATIONS 20
CONVERGE E=1.0E-3 GRAD=0.0005
STEP RAD=0.3 ANGLE=5.0
DIIS N=5 OK=0.1 CYC=3
END
XC
LDA VWN
GGA POSTSCF Becke Perdew
End
Integration 3.0
SCF
Iterations 60
Converge 1.0E-06 1.0E-6
Mixing 0.20
DIIS N=10 OK=0.500 CX=5.00 CXX=25.00 BFAC=0.00
End
End Input
In the above example, the geometry was defined with Cartesian coordinates and the geometry optimization was also done in Cartesians. The same input could also have easily been defined with a Z-matrix in the ATOMS key block.
Example B: Pd+ -Ethene pi-complexation Linear Transit¶
This example is more complex to demonstrate some problems that one might encounter in a more advanced problem. For instance, the simulation will involve the customization of the standard Tripos force field, the use of dummy atoms in the MM region and QM region and the use of constraints. Figure 5-3a depicts the system that we intend to simulate. More specifically we wish to determine the reaction profile of removing the olefinic substrate from the metal center of Pd+ -phosphino-ferrocenyl-pyrazole complex. For this purpose we wish to perform a linear transit run with ADF, whereby the distance between the metal center and the midpoint of the olefinic carbons is used as a reaction coordinate. The linear transit geometry optimization will be done in internal coordinates. This reaction coordinate is shown as the arrow line in Figure 5-3. This example originates from our research on similar neutral bis-trichlorosilyl compounds. The system has been changed slightly to introduce additional technical considerations when using dummy atoms and linear transit calculations. The QM/MM calculations of these related compounds reveal that the approximations introduced in this system are quite reasonable. Also, in terms of predicting the geometry of this class of complexes, the QM/MM method performs exceptionally well.

** a b**
Figure 5-3 Example system Pd+ -ethene pi-complex. a) Full system, with linear transit coordinate indicated by the arrow line. The QM/MM boundary is shown as the dotted polygon with the QM region residing inside. The ‘LI’ atoms are denoted with the asterisks. b) The QM model system with the capping hydrogen atoms depicted in bold.
Step 1. Partitioning the System and the Model QM system.¶
First one must decide where to partition the system into QM and MM regions. For this example, we have decide to partition the system as illustrated in Figure 53b, whereby the QM region is contained in the dotted polygon. The corresponding QM model system, for which the electronic structure calculation will be performed, is depicted in Figure 5-3b. In the model QM system the link atoms have been replaced by capping hydrogen atoms. Notice that the QM/MM boundary cuts through the cyclopentadienyl ring of the ferrocenyl ligand. Based on experimental studies of this complex, it is assumed that the ferrocenyl ligand acts only as a spectator ligand and can be modeled effectively on a steric basis only. Using an olefinic group will approximate the sp2 hybridization of the Cp rings. Here, special care must be taken to preserver the structural features of the Cp ring. For example the C-C bond distance in the ferrocenyl ligand is approximately 1.45 Ang whereas it is about 1.34 Ang in an olefin. This will be elaborated on later. The replacement of the phenyl phosphine in the real system by hydrogen phosphine will have some consequences due to the different electronic properties of the substituents. It is known that the phenyl substitution on the phosphine is more electron withdrawing than the hydrogen substituent. The replacement of the phenyl phosphine with hydrogen phosphine will result in a contraction of the Pd-P bond in the QM model system and hence the Pd-P bond will be too short in our QM/MM model.
Step 2. Labeling of Atoms¶
Once a partitioning of the system has been established, one needs to label each of the atoms in the full system as QM, LI or MM atom type. For the example system, all atoms contained within the dotted polygon in Figure 5-3a are ‘QM’ atoms. All atoms that are marked with asterisks in Figure 5-3a are ‘LI’ atoms and finally the remaining atoms outside the dotted polygon are ‘MM’ atoms. The dummy atom that we need in order to define our reaction coordinate is designated a QM atom. This dummy atom will be made to lie midway between the two olefinic carbon atoms of the ethene moiety. It is important to realize that the linear transit constraint cannot involve any MM atoms. Two dummy atoms representing the center of the Cp rings of the ferrocenyl ligand will also be introduced. They we be part of the MM subsystem.
Step 3. Renumbering of Atoms¶
It will be re-emphasize that there is a strict rule concerning the ordering of the atoms, based on their QM, MM, or LI atom type designation. All QM and LI atoms must come before any MM atoms. This rule also applies to the dummy atoms. All atoms in the QM model system shown in Figure 5-3b and their equivalent LI atoms in the real system must come first. Given below are the Cartesian coordinates of the initial geometry with the atoms renumbered. Although the optimization will be performed in internal coordinates, this is a complex example, and it might help the reader to examine the 3D structure of the complex with their favorite molecule viewer.
Pd 0.00000 0.00000 0.00000
N 2.18381 0.00000 0.00000
P -0.19353 2.33087 0.00000
Si -2.09382 -0.72920 0.40993
Cl -3.11786 -1.66043 -1.19030
Cl -3.49847 0.77293 1.00006
Cl -2.26795 -2.09058 2.02296
C 1.00751 3.35326 -0.90266
C 2.19320 2.92863 -1.63738
C 2.55933 1.49397 -1.90948
N 3.04680 0.78384 -0.70880
C 4.30216 0.71267 -0.18548
C 4.25805 -0.16196 0.88628
C 2.91893 -0.57760 0.96569
Xx 0.74788 -1.69468 -1.67891
C 1.00986 -2.06361 -1.18981
C 0.48590 -1.32574 -2.16801
H 0.42486 -2.82737 -0.67948
H 2.04440 -1.93825 -0.86750
H 1.06712 -0.55943 -2.68284
H -0.54392 -1.46716 -2.49409
C -0.02313 3.09172 1.69751
C -1.80681 3.04317 -0.56800
C 1.06326 4.78500 -0.83878
C 2.90531 4.10963 -2.00851
H 1.82015 0.84892 -1.41063
C 2.56571 1.10455 -3.38246
H 5.13150 1.27389 -0.59600
H 5.08293 -0.44584 1.52605
C 2.28008 -1.58274 1.88382
Fe 1.04114 4.20972 -2.75447
H 3.29128 1.69565 -3.95722
H 2.82573 0.03883 -3.48724
H 1.57260 1.26129 -3.82619
C 2.23262 5.27491 -1.51096
H 3.82950 4.13635 -2.57654
H 2.53722 6.31008 -1.62736
H 0.35288 5.41923 -0.32204
C -0.36634 3.33949 -3.93243
C -0.80398 4.62382 -3.46691
C 0.14944 5.60144 -3.90508
C 1.17457 4.92275 -4.64218
C 0.85084 3.52607 -4.66695
H 1.38331 2.75840 -5.21594
H -0.88905 2.39926 -3.80335
H -1.70862 4.82843 -2.90699
H 0.10418 6.66814 -3.70967
H 2.03573 5.38561 -5.11158
C -2.52881 2.31699 -1.52071
C -3.74190 2.80302 -2.01422
C -4.24163 4.02394 -1.55340
C -3.52216 4.75609 -0.60549
C -2.30668 4.26891 -0.11529
H -2.14465 1.36750 -1.87729
H -4.29443 2.23258 -2.75379
H -5.18572 4.40286 -1.93263
H -3.90891 5.70542 -0.24710
H -1.76468 4.85404 0.61704
C -1.11030 3.12908 2.57588
C -0.97634 3.68572 3.85061
C 0.25426 4.20707 4.25852
C 1.35020 4.15676 3.39277
C 1.21425 3.58782 2.12359
H -2.06730 2.72549 2.27375
H -1.82678 3.71355 4.52356
H 0.35921 4.64813 5.24539
H 2.30826 4.55806 3.70733
H 2.08227 3.52803 1.47710
C 2.71278 -2.91472 1.84328
C 2.09262 -3.86832 2.66014
C 1.02394 -3.51226 3.48850
C 0.62539 -2.17333 3.54762
C 1.26348 -1.20208 2.77018
C 3.85077 -3.34061 0.90256
H 2.44054 -4.89548 2.65117
C 0.28665 -4.56067 4.32981
H -0.18696 -1.88618 4.20708
C 0.83254 0.25922 2.93922
H 4.81150 -3.02160 1.33267
H 3.72469 -2.87842 -0.08835
H 3.87528 -4.43158 0.76399
H 1.55468 0.95128 2.48757
H 0.75353 0.50536 4.00860
H -0.15276 0.39937 2.47265
H -0.79950 -4.40165 4.25848
H 0.51050 -5.58360 3.99174
H 0.59686 -4.45719 5.38063
Xx 1.88038 4.09028 -1.37966
Xx 0.20091 4.40271 -4.12271
Step 4. Z-matrix and constraints¶
This simulation will be a linear transit simulation where the reaction coordinate will be the distance from the Pd center to the midpoint of the complexed ethene molecule. In order to use midpoint of the ethene molecule as the reaction coordinate we must use a dummy atom to define the midpoint and a few constraints to maintain the dummy atom at the midpoint. The dummy atom is atom number 15 and the two carbon atoms of the ethene moiety are atoms 16 and 17. Each of the two carbons of the ethene will be ‘bonded’ to the midpoint using the same (free) bond distance variable ‘B15’. This will ensure that the midpoint dummy atom is always equidistant to each ethene carbon. To ensure that the midpoint dummy atom always lies along the C-C bond vector the dihedral variable ‘D14’ will be constrained to 180 degrees. Finally, to prevent C-C bond distance of olefinic group used to model the ferrocenyl ligand to revert to its natural bond length of approximately 1.34 Ang, the C-C distance (B8) will be constrained to 1.45 Ang. This is the distance found in the C-C bond distance in the ferrocenyl ligand. One might be concerned about the internal coordinate definition of the cyclopentadienyl rings of the ferrocenyl ligand. Since the ferrocenyl ligand is part of the MM region, the Z-matrix will be used only to construct the initial geometry. From there the molecular mechanics borderleft takes over and where the optimization is done in Cartesian coordinates. For MM atoms it is important that care be taken when defining the connection table.
ATOMS internal
Pd 0 0 0 0 0 0
N 1 0 0 B1 0 0
P 1 2 0 B2 A1 0
Si 1 2 3 B3 A2 D1
Cl 4 1 2 B4 A3 D2
Cl 4 1 5 B5 A4 D3
Cl 4 1 5 B6 A5 D4
C 3 1 2 B7 A6 D5
C 8 3 1 B8 A7 D6
C 9 8 3 B9 A8 D7
N 10 9 8 B10 A9 D8
C 11 2 1 B11 A10 D9
C 12 11 2 B12 A11 D10
C 2 1 11 B13 A12 D11
XX 1 2 3 B14 A13 D12
C 15 1 2 B15 A14 D13
C 15 1 16 B15 A15 D14
H 16 15 1 B17 A16 D15
H 16 17 18 B18 A17 D16
H 17 16 18 B19 A18 D17
H 17 16 18 B20 A19 D18
C 3 1 8 B21 A20 D19
C 3 1 8 B22 A21 D20
C 8 3 9 B23 A22 D21
C 9 8 10 B24 A23 D22
H 10 9 8 B25 A24 D23
C 10 9 26 B26 A25 D24
H 12 11 13 B27 A26 D25
H 13 12 11 B28 A27 D26
C 14 2 1 B29 A28 D27
Fe 24 8 3 B30 A29 D28
H 27 10 9 B31 A30 D29
H 27 10 32 B32 A31 D30
H 27 10 32 B33 A32 D31
C 25 9 8 B34 A33 D32
H 25 9 35 B35 A34 D33
H 35 25 9 B36 A35 D34
H 24 8 31 B37 A36 D35
C 31 24 8 B38 A37 D36
C 39 31 24 B39 A38 D37
C 40 39 31 B40 A39 D38
C 41 40 39 B41 A40 D39
C 39 31 40 B42 A41 D40
H 43 39 31 B43 A42 D41
H 39 31 40 B44 A43 D42
H 40 39 41 B45 A44 D43
H 41 40 42 B46 A45 D44
H 42 41 40 B47 A46 D45
C 23 3 1 B48 A47 D46
C 49 23 3 B49 A48 D47
C 50 49 23 B50 A49 D48
C 51 50 49 B51 A50 D49
C 52 51 50 B52 A51 D50
H 49 23 50 B53 A52 D51
H 50 49 51 B54 A53 D52
H 51 50 52 B55 A54 D53
H 52 51 53 B56 A55 D54
H 53 52 51 B57 A56 D55
C 22 3 1 B58 A57 D56
C 59 22 3 B59 A58 D57
C 60 59 22 B60 A59 D58
C 61 60 59 B61 A60 D59
C 62 61 60 B62 A61 D60
H 59 22 60 B63 A62 D61
H 60 59 61 B64 A63 D62
H 61 60 62 B65 A64 D63
H 62 61 63 B66 A65 D64
H 63 62 61 B67 A66 D65
C 30 14 2 B68 A67 D66
C 69 30 14 B69 A68 D67
C 70 69 30 B70 A69 D68
C 71 70 69 B71 A70 D69
C 72 71 70 B72 A71 D70
C 69 30 70 B73 A72 D71
H 70 69 71 B74 A73 D72
C 71 70 72 B75 A74 D73
H 72 71 73 B76 A75 D74
C 73 72 71 B77 A76 D75
H 74 69 30 B78 A77 D76
H 74 69 79 B79 A78 D77
H 74 69 79 B80 A79 D78
H 78 73 72 B81 A80 D79
H 78 73 82 B82 A81 D80
H 78 73 82 B83 A82 D81
H 76 71 70 B84 A83 D82
H 76 71 85 B85 A84 D83
H 76 71 85 B86 A85 D84
XX 24 8 31 B87 A86 D85
XX 41 40 42 B88 A87 D86
END
GEOVAR
B1=2.18381
B2=2.33889
B3=2.25474
B4=2.11579
B5=2.13955
B6=2.11791
B7=1.81730
B8=1.45807 F
B9=1.50544
B10=1.47768
B11=1.36193
B12=1.38405
B13=1.34409
B14=2.50000 5.000
B15=0.66631
B17=1.08903
B18=1.09082
B19=1.09092
B20=1.08943
B21=1.86801
B22=1.85275
B23=1.43426
B24=1.42814
B25=1.10060
B26=1.52360
B27=1.08226
B28=1.08182
B29=1.50380
B30=2.00032
B31=1.09827
B32=1.10198
B33=1.09896
B34=1.43456
B35=1.08513
B36=1.08532
B37=1.08347
B38=2.03122
B39=1.43448
B40=1.43413
B41=1.43347
B42=1.43383
B43=1.08361
B44=1.08347
B45=1.08341
B46=1.08540
B47=1.08451
B48=1.39867
B49=1.39691
B50=1.39740
B51=1.39722
B52=1.39822
B53=1.08455
B54=1.08520
B55=1.08569
B56=1.08594
B57=1.08280
B58=1.39816
B59=1.39740
B60=1.39734
B61=1.39754
B62=1.39750
B63=1.08166
B64=1.08484
B65=1.08604
B66=1.08530
B67=1.08396
B68=1.40109
B69=1.40042
B70=1.39823
B71=1.39824
B72=1.39818
B73=1.53667
B74=1.08452
B75=1.53315
B76=1.08501
B77=1.53287
B78=1.09990
B79=1.10064
B80=1.10001
B81=1.09746
B82=1.10018
B83=1.09915
B84=1.10005
B85=1.10036
B86=1.10053
B87=1.20119
B88=1.21941
A1=94.7463
A2=158.2223
A3=117.0192
A4=115.6423
A5=115.0054
A6=120.3960
A7=128.6001
A8=124.4982
A9=113.0381
A10=110.6944
A11=107.5291
A12=123.1566
A13=72.5934
A14=90.0001
A15=90.0001
A16=121.4060
A17=121.7463
A18=121.7273
A19=121.2990
A20=113.4758
A21=117.0770
A22=124.4140
A23=107.1936
A24=108.2564
A25=114.7923
A26=122.2418
A27=126.8321
A28=119.5733
A29=70.7204
A30=111.7865
A31=109.8825
A32=110.4946
A33=110.3514
A34=125.5260
A35=127.2980
A36=125.4579
A37=133.3349
A38=68.8766
A39=107.9605
A40=108.0463
A41=69.7028
A42=125.7664
A43=129.5676
A44=126.2672
A45=125.9595
A46=126.0404
A47=117.3031
A48=120.5489
A49=119.8703
A50=119.8287
A51=120.1922
A52=119.7123
A53=120.0017
A54=120.0478
A55=119.9075
A56=118.9447
A57=120.7197
A58=120.6090
A59=119.9563
A60=119.7586
A61=120.0640
A62=120.1660
A63=120.0615
A64=120.1253
A65=119.9602
A66=119.0914
A67=119.1156
A68=119.5849
A69=120.7057
A70=119.1275
A71=120.7701
A72=120.6568
A73=119.8697
A74=121.2330
A75=119.5074
A76=118.2033
A77=109.0914
A78=110.4834
A79=111.6228
A80=111.7639
A81=109.9169
A82=109.0716
A83=109.9009
A84=111.6715
A85=108.9122
A86=55.1829
A87=54.0180
D1=-150.6568
D2=-100.8347
D3=-119.5809
D4=123.4778
D5=35.1598
D6=-2.9958
D7=7.2171
D8=-72.9107
D9=164.5027
D10=0.9220
D11=163.0063
D12=135.2677
D13=65.6662
D14=180.0000 F
D15=90.0000
D16=179.5031
D17=-179.9904
D18=-0.4168
D19=-117.3520
D20=124.7005
D21=-169.6021
D22=-179.2713
D23=-3.3919
D24=119.3500
D25=179.9891
D26=-179.9514
D27=17.4102
D28=-129.2972
D29=60.6432
D30=120.3390
D31=-120.3043
D32=1.2969
D33=179.4155
D34=179.5397
D35=123.6746
D36=68.3005
D37=62.0893
D38=58.4426
D39=0.0736
D40=119.7199
D41=127.1496
D42=-120.1862
D43=178.4186
D44=-179.8093
D45=179.5019
D46=-30.2454
D47=-178.8402
D48=-0.0598
D49=0.3012
D50=-0.1575
D51=-179.8476
D52=-179.9824
D53=179.8951
D54=-179.8926
D55=179.8489
D56=-81.7949
D57=179.3002
D58=-0.2287
D59=-0.6445
D60=0.0510
D61=-179.9960
D62=179.9913
D63=-179.8879
D64=-179.8700
D65=-178.0390
D66=114.6178
D67=-177.7647
D68=1.7082
D69=-3.4460
D70=1.3564
D71=179.4189
D72=179.8505
D73=-179.9397
D74=-179.8352
D75=-176.6101
D76=76.9134
D77=-120.1615
D78=119.9038
D79=165.3767
D80=-119.6911
D81=121.2928
D82=-135.8162
D83=120.3122
D84=-119.4211
D85=-58.7928
D86=0.0762
END
Step 5. Connection Table, MM force field atom-types and Force Field Modification¶
In order to construct a molecular mechanics potential, the program needs to know the connectivity of the molecular system and the molecular mechanics force field atom-type designations. In this example we are using the Tripos or Sybyl force field. The Tripos force field does not support either Pd or ferrocenyl ligands, so we need to modify the standard force field file to handle these groups. Modification of a molecular mechanics force field without re-parameterization of the force field may not always be appropriate. However, in this case sort of ‘ad hoc’ additions to the Tripos force field can be justified. For Pd, all of the principle interactions will be contained within the QM region and only weak non-bonded interactions involving Pd will be approximated by the molecular mechanics potential. The ferrocenyl ligand is assumed to act as a spectator ligand and therefore it is adequate to simply attain the approximate structure of the complex with the molecular mechanics potential.
In the Tripos force field, the nitrogen atoms of the pyrazole ring should be assigned the ‘N_2’ atom-type; the P atom of the phosphine should be assigned the ‘P_3’ atom-type. The Cl, H and Si atoms are given the ‘Cl’, ‘H’, and ‘Si’ atom types respectively. The carbon atoms of the phenyl substituents are given the ‘C_ar’ atom-type, while the sp3 hybridized carbon atoms are given the ‘C_3’ atom-type.
Connections involving the dummy atom defining the midpoint of the ethene molecule are really not needed since this atom is contained within the QM region.
For the ferrocenyl ligand the ferrocene force field of Bosnich and coworkers will be used. Four new MM atom types will be introduced, C_cp, H_cp and CEN, representing the carbon, hydrogen and centroid of the cyclopentadienyl rings, respectively and Fe. In the connection table, the C_cp atoms will be bonded to the centroid and not the Fe center. The only two bonds made to the Fe center will be to the (two) central dummy atoms of the Cp rings. In making a connection between the C_cp atom and the centroid, a direct bond will is made to a QM atom and a MM atom. A warning will be issued during the run but as long as the ‘WARNING_LEVEL’ flag is set to 1 the job will continue. In this case the link bond between the C_cp atom and the centroid does not need to be mediated by a capping atom. This bond is used only for the construction of the MM potential for the ferrocenyl ligand. Special bond stretching, bending, torsion and out-of-plane potentials need to be added to the force field file. For the most part these parameters are taken from the Bosnich Ferrocene force field. For example for the bond stretches, the following potentials need to be added to the force field.
# Parameters added for Pd - ethene complex
C_cp C_cp 1 1400.00 1.434 From the Bosnich ferrocene force field.
C_cp H_cp 1 692.00 1.085 Bosnich
CEN Fe 1 600.00 1.617 Bosnich
CEN C_cp 1 600.00 1.220 Bosnich
The force field file is simply a text file and so the above section needs to be added to the ‘BONDS’ key block between the two ‘=======’ separator lines. Bond potentials need to be defined between the centroid of the Cp rings with the Fe center and the carbon atoms.
For the angle and torsion terms, the additions are somewhat more complex. The following angle potential terms need to be introduced.
# Parameters added for Pd - ethene complex
C_cp C_cp C_cp 1 78.80 126.0 Bosnich
C_cp C_cp H_cp 1 78.80 126.0 Bosnich
CEN C_cp C_cp 1 0.00 0.0 no potential
CEN C_cp H_cp 1 0.00 0.0 no potential
C_cp CEN C_cp 1 0.00 0.0 no potential
C_cp CEN Fe 1 100.00 90.0 Bosnich
CEN Fe CEN 1 100.00 180.0 Bosnich
CEN C_cp P_3 1 0.00 0.0 no potential
CEN C_cp C_3 1 0.00 0.0 no potential
Any angle potentials involving the Cp centroid and any atoms outside of the ferrocenyl ligand have been set to zero since the centroid was only a construct for the optimization of the ferrocenyl ligand. For the torsions, the following potentials have been added to the standard Tripos force field.
# Parameters added for Pd - ethene complex
P_3 C_cp C_cp C_cp 2 2.0000 -2.0 Sybyl *-C_ar-C_ar-* aromatic bond
P_3 C_cp C_cp H_cp 2 2.0000 -2.0 Sybyl *-C_ar-C_ar-* aromatic bond
C_3 C_cp C_cp H_cp 2 2.0000 -2.0 Sybyl *-C_ar-C_ar-* aromatic bond
H_cp C_cp C_cp H_cp 2 2.0000 -2.0 Sybyl *-C_ar-C_ar-* aromatic bond
* C_cp C_cp C_cp 2 2.3500 -2.0 same as SYBYL * C_ar C_ar C_ar
* Fe CEN * 0 0.0000 -2.0 no potential involving centroid
* C_cp CEN * 0 0.0000 0.0 no potential involving centroid
* C_cp C_cp CEN 0 0.0000 0.0 no potential involving centroid
Pd P_3 C_cp CEN 0 0.0000 0.0 no potential involving centroid
CEN C_cp P_3 C_ar 0 0.0000 0.0 no potential involving centroid
N_2 C_3 C_cp CEN 0 0.0000 0.0 no potential involving centroid
CEN C_cp C_3 H 0 0.0000 0.0 no potential involving centroid
CEN C_cp C_3 C_3 0 0.0000 0.0 no potential involving centroid
Here any torsional potentials involving the Centroid atom of the ferrocenyl ligand were set to zero. Torsional potentials involving atoms outside of the ferrocenyl ligand and having the C_cp-C_cp atoms central atom pair, these potentials were equated with those of the Tripos ‘ *-C_ar C_ar - * ‘ torsional potentials. Again, these somewhat arbitrary choices for the MM potentials involving the ferrocenyl ligand are justified by the fact that the ferrocenyl ligand acts only as a spectator group.
The van der Waals parameters used for the five new atoms types, Pd, Fe, CEN, C_cp and H_cp were taken from either existing Tripos van der Waals parameters of similar atom-types or they were taken from Rappe’s UFF (Universal Force Field). They are given below with their origins provided in the ‘NOTES’ column.
# Parameters added for Pd - ethene complex
C_cp 0.1070 3.4000 12.00 same as Tripos C_ar
Fe 0.0130 2.9120 12.00 UFF92 Fe6+2
Pd 0.0480 2.8990 12.00 UFF92 Pd4+2
CEN 0.0000 1.0000 12.00 zero
H_cp 0.0420 3.0000 12.00 same as Tripos H
Now that the addition of the new MM potentials and atom-types has been discussed, the ‘MM_CONNECTION_TABLE’ subkey block is given below. In practice, one typically constructs the input first, and then runs the program to see what force field potentials/parameters are missing. If any force field parameters are missing in the force field file, the ADF QM/MM program will print all missing potentials that need to be defined in the force field and then stop.
MM_CONNECTION_TABLE
1 Pd QM 2 3 4 0 0 0
2 N_2 QM 1 11 14 0 0 0
3 P_3 QM 1 8 22 23 0 0
4 Si QM 1 5 6 7 0 0
5 Cl QM 4 0 0 0 0 0
6 Cl QM 4 0 0 0 0 0
7 Cl QM 4 0 0 0 0 0
8 C_cp QM 3 9 24 88 0 0
9 C_cp QM 8 10 25 88 0 0
10 C_3 QM 9 11 26 27 0 0
11 N_2 QM 2 10 12 0 0 0
12 C_ar QM 11 13 28 0 0 0
13 C_ar QM 12 14 29 0 0 0
14 C_ar QM 2 13 30 0 0 0
15 XX QM 16 17 1 0 0 0
16 C_2 QM 15 17 18 19 0 0
17 C_2 QM 15 16 20 21 0 0
18 H QM 16 0 0 0 0 0
19 H QM 16 0 0 0 0 0
20 H QM 17 0 0 0 0 0
21 H QM 17 0 0 0 0 0
22 C_ar LI 3 59 63 0 0 0
23 C_ar LI 3 49 53 0 0 0
24 C_cp LI 8 35 38 88 0 0
25 C_cp LI 9 35 36 88 0 0
26 H QM 10 0 0 0 0 0
27 C_3 LI 10 32 33 34 0 0
28 H QM 12 0 0 0 0 0
29 H QM 13 0 0 0 0 0
30 C_ar LI 14 69 73 0 0 0
31 Fe MM 88 89 0 0 0 0
32 H MM 27 0 0 0 0 0
33 H MM 27 0 0 0 0 0
34 H MM 27 0 0 0 0 0
35 C_cp MM 24 25 37 88 0 0
36 H_cp MM 25 0 0 0 0 0
37 H_cp MM 35 0 0 0 0 0
38 H_cp MM 24 0 0 0 0 0
39 C_cp MM 40 43 45 89 0 0
40 C_cp MM 39 41 46 89 0 0
41 C_cp MM 40 42 47 89 0 0
42 C_cp MM 41 43 48 89 0 0
43 C_cp MM 39 42 44 89 0 0
44 H_cp MM 43 0 0 0 0 0
45 H_cp MM 39 0 0 0 0 0
46 H_cp MM 40 0 0 0 0 0
47 H_cp MM 41 0 0 0 0 0
48 H_cp MM 42 0 0 0 0 0
49 C_ar MM 23 50 54 0 0 0
50 C_ar MM 49 51 55 0 0 0
51 C_ar MM 50 52 56 0 0 0
52 C_ar MM 51 53 57 0 0 0
53 C_ar MM 23 52 58 0 0 0
54 H MM 49 0 0 0 0 0
55 H MM 50 0 0 0 0 0
56 H MM 51 0 0 0 0 0
57 H MM 52 0 0 0 0 0
58 H MM 53 0 0 0 0 0
59 C_ar MM 22 60 64 0 0 0
60 C_ar MM 59 61 65 0 0 0
61 C_ar MM 60 62 66 0 0 0
62 C_ar MM 61 63 67 0 0 0
63 C_ar MM 22 62 68 0 0 0
64 H MM 59 0 0 0 0 0
65 H MM 60 0 0 0 0 0
66 H MM 61 0 0 0 0 0
67 H MM 62 0 0 0 0 0
68 H MM 63 0 0 0 0 0
69 C_ar MM 30 70 74 0 0 0
70 C_ar MM 69 71 75 0 0 0
71 C_ar MM 70 72 76 0 0 0
72 C_ar MM 71 73 77 0 0 0
73 C_ar MM 30 72 78 0 0 0
74 C_3 MM 69 79 80 81 0 0
75 H MM 70 0 0 0 0 0
76 C_3 MM 71 85 86 87 0 0
77 H MM 72 0 0 0 0 0
78 C_3 MM 73 82 83 84 0 0
79 H MM 74 0 0 0 0 0
80 H MM 74 0 0 0 0 0
81 H MM 74 0 0 0 0 0
82 H MM 78 0 0 0 0 0
83 H MM 78 0 0 0 0 0
84 H MM 78 0 0 0 0 0
85 H MM 76 0 0 0 0 0
86 H MM 76 0 0 0 0 0
87 H MM 76 0 0 0 0 0
88 CEN MM 8 9 24 25 35 31
89 CEN MM 39 40 41 42 43 31
SUBEND
Step 6. LINK_BONDS¶
In this example there are 6 link bonds as depicted in Figure 5-3a. The link bond parameters a for each of these bonds will be determined by comparing bond lengths in the X-ray structure of a similar bis-trichlorosilyl Pd complex with that of the calculated pure QM gas-phase structure of the QM model system. As shown in Figure 5-3b all link bonds will be capped with Hydrogen atoms. Although the MM connection table defines direct bonds between atoms 8 and 9 (C Cp atoms) with the centroid of the Cp rings, they are not mediated by capping atoms. In other words the Cp centroid is defined as a MM atom-type not a LI atom type. Therefore, no link parameters are necessary for those two bonds.
LINK_BONDS
:: ---------- ------ ---
:: atoms alpha dummy
:: ---------- ------ ---
22 - 3 1.2990 H
23 - 3 1.2990 H
24 - 8 1.3200 H
25 - 9 1.3200 H
27 - 10 1.3710 H
30 - 14 1.3800 H
:: ---------- ------ ---
SUBEND
Step 7. CHARGES¶
In this example, the Pd center has a formal positive charge. In order to obtain the proper electronic structure in the calculation of the QM model system, we must define in the ADF input a formal positive charge with the CHARGE keyword.
CHARGE 1 ! note this is in the main ADF input
The original Tripos force field was parameterized without explicit electrostatic terms. Thus, we will use this convention and turn off the electrostatic coupling between the QM and MM regions using the ELSTAT_COUPLING_MODEL keyword in the QMMM key block.
ELSTAT_COUPLING_MODEL=0
Although this may seem like a dubious choice, experience with organometallic complexes has shown that this is a good approximation. For other types of molecular systems, namely amino and nucleic acids it is not a good choice to turn off the electrostatic coupling between the QM and MM regions. Furthermore, force fields designed for this biochemical species almost always have charges included in the parameterization process.
Step 9. Putting it all together: The whole ADF QM/MM input.¶
The whole ADF QM/MM input for the sample system is given below. Some comments are provided in bold. Additionally, some lengthy sections have been omitted that have already been given in full above.
TITLE Complex force field Example of Pd+-Ethene complex.
NOPRINT SFO
Fragments
Pd T21.Pd.3d.rel
C T21.C.1s.rel
Si T21.Si.2p.rel
Cl T21.Cl.2p.rel
H T21.H.rel
N T21.N.1s.rel
P T21.P.2p.rel
End
RELATIVISTIC SCALAR ZORA
SYMMETRY NOSYM
CHARGE 1 ! CHARGE is defined from the QM model system
GEOMETRY
LINEAR TRANSIT 4
ITERATIONS 2
HESSUPD BFGS
CONVERGE E=2.0E-3 GRAD=0.002
DIIS N=5 OK=0.005 CYC=2
END
XC
LDA VWN
GGA Becke Perdew
End
Integration 3.0 3.0
SCF
Iterations 60
Converge 1.0E-06 1.0E-06
Mixing 0.20
DIIS N=10 OK=0.500 CX=5.00 CXX=25.00 BFAC=0.00
LShift 0.00
End
QMMM
FORCE_FIELD_FILE sybyl.ff
OUTPUT_LEVEL=1
WARNING_LEVEL=-1
ELSTAT_COUPLING_MODEL=0
MM_CONNECTION_TABLE
! SAME AS IN ABOVE
SUBEND
LINK_BONDS
:: ---------- ------ ---
:: atoms alpha dummy
:: ---------- ------ ---
22 - 3 1.2990 H
23 - 3 1.2990 H
24 - 8 1.3200 H
25 - 9 1.3200 H
27 - 10 1.3710 H
30 - 14 1.3800 H
:: ---------- ------ ---
SUBEND
END
ATOMS internal
! SAME AS IN ABOVE
END
GEOVAR
! SAME AS IN ABOVE
END
END INPUT