5.3. Refitting HF Charges with the ACKS2 Model¶
This example demonstrates the use of the ParAMS Main Script and will assume that the reader is already familiar with this section. Following the paper Atom-condensed Kohn-Sham DFT approximated to second order (ACKS2) by Verstraelen et al., we will show how to refit the ReaxFF charges model, specifically for the atoms \(\mathrm{H}\) and \(\mathrm{F}\).
Looking inside the $AMSHOME/scripting/scm/params/examples/HF_ACKS2 directory, you should find the following structure:
HF_ACKS2
├── inputs
│ ├── CHONSMgPNaFBLi-e.ff
│ └── reference.dat
├── params.conf.py
└── run.py
Inside the inputs folder is the force field that will be parameterized. The file reference.dat is holding the reference data that we will be fitting to: Partial charge at the Hydrogen as a function of the H-F distance. The reference data has been extracted from the publication’s Fig. 3a.
5.3.1. The Config File¶
As explained in the main script section, all further settings regarding a parameterization are saved in the params.conf.py file. Taking a look at the first lines, we will see the following:
### REQUIRED: Construct an instance of an interface to one of the different
### parameterized methods.
interface = ReaxParams('inputs/CHONSMgPNaFBLi-e.ff')
# We want to parameterize only the EEM model for H and F
active_params = [
'F:gamma_i;;24;;gammaEEM, EEM shielding',
'F:chi_i;;24,25;;EEM electronegativity',
'F:eta_i;;24,25;;EEM hardness',
'F:C_i;;25;;Atomic softness cutoff parameter',
'H:gamma_i;;24;;gammaEEM, EEM shielding',
'H:chi_i;;24,25;;EEM electronegativity',
'H:eta_i;;24,25;;EEM hardness',
'H:C_i;;25;;Atomic softness cutoff parameter',
'X_soft;;25;;ACKS2 softness parameter',
]
interface.is_active = len(interface)*[False] # Switch all parameters off
for name in active_params: # Switch the names above back on
interface[name].is_active = True
While interface variable is simply pointing to the force field file,
note the following lines.
From the name of the force field it is clear that there are many
more atomic parameters present than just the ones for Hydrogen and Fluorine.
In this case, however, we explicitly want to fit only the ones that are related to the two atoms,
and more specifically to the ACKS2 model.
We therefore only select the parameters that are included in the active_params variable
(for naming conventions, see the ReaxFF section).
The last three lines are making use of the parameter interfaces API to conveniently switch relevant
parameters on, and the remaining ones off (for more information about the functionality of the parameter interfaces,
see the respective Parameter Interfaces section).
5.3.2. Preparing the YAML Files from Input¶
As noted in the main script section, the only additional files needed for the optimization are the two YAML files jobcollection.yaml and trainingset.yaml.
Execute the following in a terminal:
>>> ./run.py prep
Preparing files from inputs/*
Storing 'outputs/jobcollection.yaml'
Storing 'outputs/trainingset.yaml'
Preparations done!
You can now run 'params optimize'.
We encourage you to open and inspect the created YAML files in a text editor.
5.3.3. Running the Optimization¶
We have now generated a Job Collection and Data Set from the reference.dat in our inputs.
Typing params optimize will start the optimization:
>>> params optimize
Reading ParAMS config ...
eRaxFF, will enable related parameters.
Number of active parameters to optimize: 9
Loading job collection from outputs/jobcollection.yaml ... done.
Loading data set from outputs/trainingset.yaml ... done.
No reference engines used.
Checking project for consistency ...
No issues found.
Starting parameter optimization.
Initial f(x)=2.952e+00
Plotting functionality not guaranteed: Matplotlib appears to be out of date. Consider upgrading.
Initial f(x) = 1.754e-01
At CMA Iteration: 0001. Best f(x)=1.507e-01.
At CMA Iteration: 0002. Best f(x)=1.056e-01.
At CMA Iteration: 0003. Best f(x)=1.056e-01.
...
Callback: MaxIter
Fitness function value is f(x) = 8.177e-02 after 0:02:01
Parameter optimization successful!
The optimization should finish after about two minutes with the above messages, the folder structure should now be as follows:
HF_ACKS2
├── inputs
│ ├── CHONSMgPNaFBLi-e.ff
│ └── reference.dat
├── optimization << Created by `params optimize`
│ ├── trainingset_best_params
│ ├── trainingset_history.dat
│ ├── cmadata
│ ├── plams_workdir
│ └── data
├── outputs << Created by `./run.py prep`
│ ├── trainingset.yaml
│ └── jobcollection.yaml
├── params.conf.py
└── run.py
An optimized force field has been saved in optimization/trainingset_best_params. Before comparing the old and new force fields, we will take a closer look into the optimization directory.
We can check the course of the parameterization by plotting the trainingset_history.dat
Inside the optimization/ directory call params plot . --yscale log to plot the
loss function value as a function of the evaluation number:
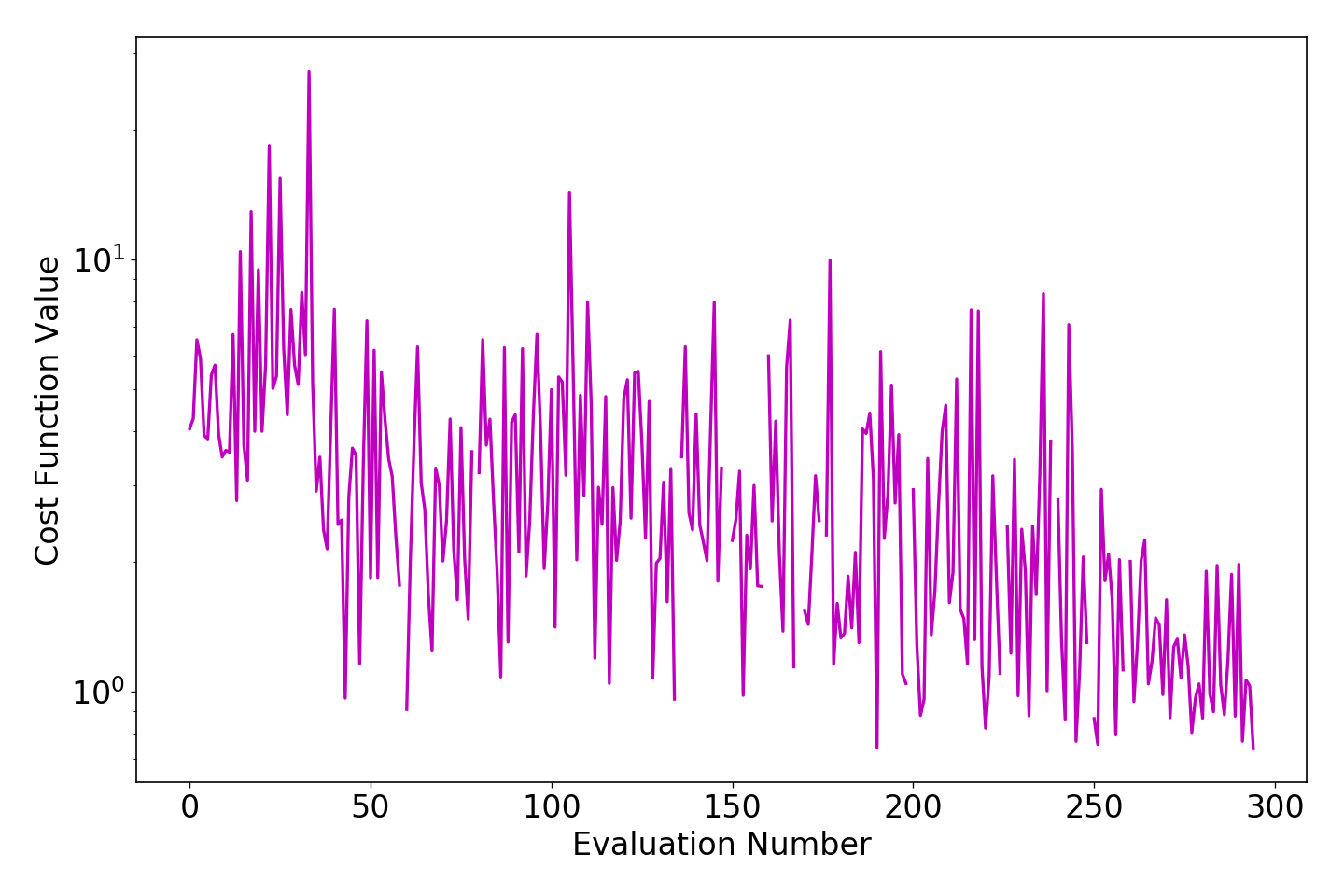
The plot shows the fitness function value for every parameter set that has been considered by the optimizer as a function of the evaluation number.
*.dat files in the optimization/data/trainingset_residuals directory contain
the residuals vector \((w/\sigma)(y-\hat{y})\)
for every improved parameter set.
In this example, \(y\) and \(\hat{y}\) are our reference and predicted charges
at the Hydrogen atom.
A histogram of every residuals file can also be created with params plot.
Generally, the closer a distribution of residuals is to zero, the better the parameters.
Residuals for the initial force field (before the start of the optimization) are saved under
data/initial_trainingset_residuals.
Finally, the optimization/data/trainingset_contributions directory contains the contributions of every data set entry to the the overall loss function value. This makes it easy to detect possible outliers. A plotted version of any contributions file looks as follows:
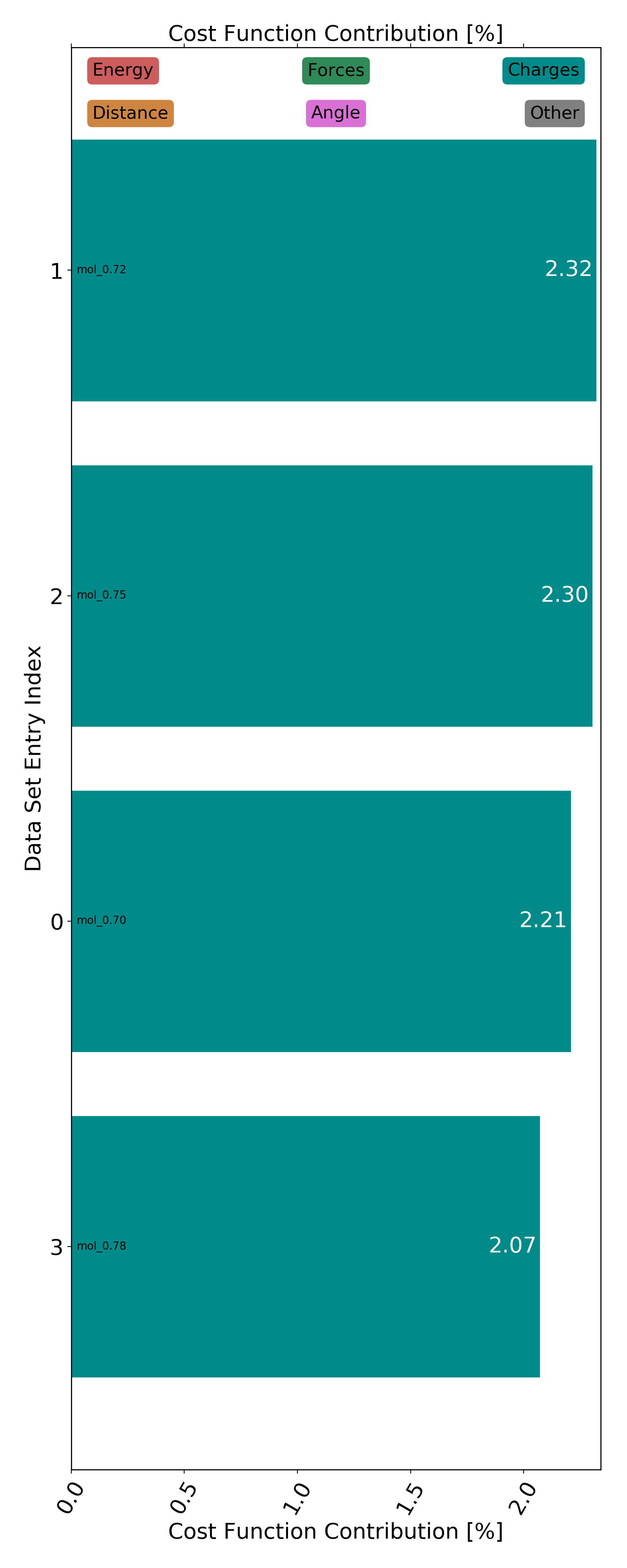
The per entry contributions of the initial force field parameters (before the start of the optimization) are saved in initial_trainingset_contributions.
5.3.4. Comparing the new parameters¶
A final comparison between old and new parameters will likely be different for every application. In this example, we provide a simple evaluation with (make sure the matplotlib module up to date):
>>> ./run.py plot
Running jobs ...
Plotting ...
Done.
This part of the script will run all structures contained in inputs/reference.dat with both, the original and optimized force fields and plot the Hydrogen partial charges as a function of the interatomic distance. The plot can be found in outputs/results.png and should look similar to the following figure.
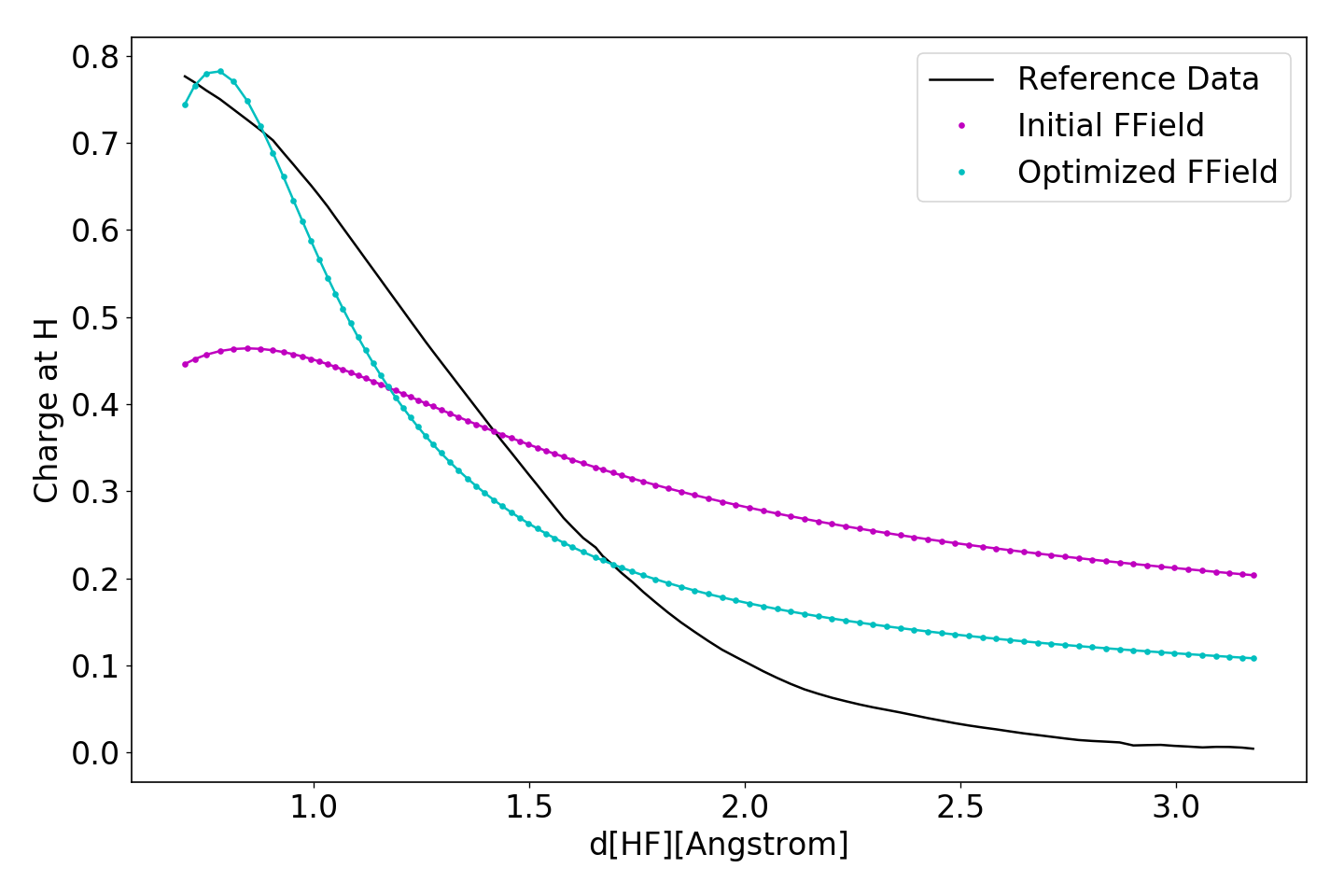
Running ./run.py clean will revert the directory to it’s original state, deleting all created files.
5.3.5. Playing with the example¶
Below are suggestions on how the existing example can be easily modified to produce different results:
Take a look at the callbacks section in the params.conf.py:
### OPTIONAL: List of callbacks to be called during the optimization.
### See the documentation for more details about callbacks.
callbacks = [
MaxIter(300), # Stop the optimization after N evaluations
# Timeout(1*60*60), # Stop the optimization after N seconds, returning the best parameters so far
# EarlyStopping(patience=150), # Stop the optimization if no improvement in the validation set aftert N evaluations
]
The example is limited to 300 evaluations. Set it to a higher number (or set a Timeout callback instead) to see if the resulting parameters can be improved further.
Take a look at the optimization_kwargs in the params.conf.py:
### See the documentation for more details about Optimization() keyword arguments.
optimization_kwargs = {
# 'loss' : 'sse', # The loss function to be evaluated.
# 'validation' : None, # Percentage of the training set to be used as validation, or another DataSet() instance
# 'batch_size' : None, # At every iteration, only compute a maximum of `batch_size` properties
# 'maxjobs' : None, # At every iteration, only run `maxjobs` random jobs and compute their properties
# 'maxjobs_shuffle' : False, # If `maxjobs`, randomly pick a new subset at every iteration if `True`.
# 'use_pipe' : True, # Use the AMSPipe interface where possible
# 'n_cores' : None, # Use N CPU cores for the execution of jobs during an optimization. Defaults to the number of physical cores
}
To allow a validation set, uncomment the respective line (also see the Optimization sections for more information about the keyword arguments).