Exciton transfer integrals¶
ADF can provide input parameters, such as exciton transfer integrals, that are needed in approximate methods that model exciton transport properties, like Förster and Marcus theory.
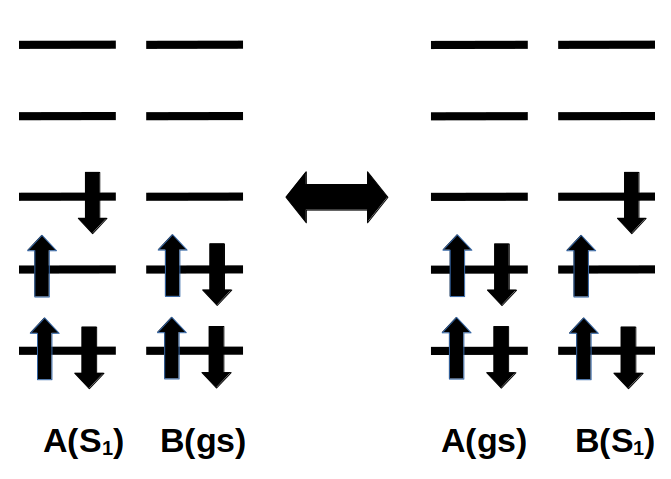
Example electron configuration for exciton hopping from A to B or back, where A and B are nearby molecules, or different parts of a complex AB. The hopping can occur from the diabatic state where A is in an excited state and B is in the ground state to the diabatic state where A is in the ground state and B is in an excited state (or back).
In theoretical models of exciton hopping, the whole system is divided into fragments, in which an exciton is localized on a fragment (diabatic state), and can hop from one fragment to another. The models require accurate values of excitonic couplings for exciton transfer (also referred to as exciton transfer integrals) and exciton site energies (energy of an exciton when it is localized at a particular molecule) as a function of the geometric conformation of adjacent molecules. Excition transfer integrals can be calculated from the energetic splitting of excitation energies in a system consisting of two adjacent molecules, also called “Davydov splitting”. ADF can also calculate exciton transfer integrals by the use of its unique fragment approach. ADF allows one to use molecular orbitals on individual molecules as a basis set in calculations on a system composed of two or more molecules. Also, ADF’s methods are applicable in cases where an orbital on one molecule couples with two or more orbitals on another molecule.
Exciton transfer integrals with FDE¶
The linear-response subsystem TDDFT code couples the monomer excitations to obtain the excited states of the total system (often denoted as coupled frozen density embedding, FDEc). It can be used, for example, to calculate singlet energy transfer integrals. See for a complete description the FDE part of the ADF manual, and particular the part about subsystem TDDFT.
Exciton transfer integrals with FOCDFT¶
FOCDFT can be used to calculate exciton transfer integrals. FOCDFT is a fragment orbital based method for constraining (integer) charges and spins on fragments. It uses the (localized) Lowdins that ADF makes to to constrain charges and spins on fragments. In practice this method works best if there are no covalent bonds or if one does not want covalent bonds between the fragments. In combination with the EXCITATIONS and EXCITONTRANSFER key one can use the localized orbitals that FOCDFT makes to calculate localized excitations on fragments (diabatic states) and calculate electronic couplings between them. The Tamm–Dancoff approximation (TDA) is required. Symmetry can be used. Because charge-transfer (CT) excitations are calculated in this method, best is to use an XC-functional that can calculate CT excitattions reasonably accurate, like, for example, range separated hybrids.
One can study, for example, singlet energy transfer (SET) or triplet energy transfer (TET) with this method.
including couplings to charge separated states, since CT states are calculated in this method.
One of the possibilities using the key ExcitonTransfer is to make purely local excitations (LEs) and purely charge-transfer (CT) excitations, if one sets the subkey ExcitonTransfer%Localize to OccupiedAndVirtual. The calculated LEs are then linear combinations of single-orbital transitions from occupied orbitals on one fragment to virtual orbitals on the same fragment. The calculated CT excitations are linear combinations of single-orbital transitions from occupied orbitals on one fragment to virtual orbitals on a different fragment. This is how excitations were analyzed in Ref. [1] using different localized orbitals than FOCDFT makes. This is not the recommended way if one uses FOCDFT localized orbitals. The FOCDFT localized occupied orbitals are not very basis set dependent. The FOCDFT localized virtual orbitals on the other hand, are much more basis set dependent. In order to make LEs and CT excitations that are not so basis set dependent a method has been implemented that combines the LEs from one fragment and CT excitations coming from the same fragment. This method will be used if one sets the subkey ExcitonTransfer%Localize to OccupiedOnly. The calculated excitations are in this case linear combinations of single-orbital transitions from occupied orbitals on one fragment to virtual orbitals on all fragments. One needs to use the Excitations%descriptors key to see whether an excitation is mostly a LE or a CT excitation.
fragments
Frag1 Frag1.results/adf.rkf
Frag2 Frag2.results/adf.rkf
end
FOCDFT
end
TDA
Excitations
descriptors
End
ExcitonTransfer
Localize OccupiedOnly
End
ExcitonTransfer
FilteredCouplings
MaxEnergy float
MaxEnergyDiff float
MinCoupling float
MinEnergy float
End
FullRun Yes/No
LocalCouplingsOnly Yes/No
Localize [OccupiedOnly | OccupiedAndVirtual]
Output [AllCouplings | FilteredCouplings | AllAndFilteredCouplings]
SecondOrder Yes/No
UseRose Yes/No
End
ExcitonTransfer- Type:
Block
- Description:
Block key for exciton transfer integrals with ROSE or FOCDFT.
FilteredCouplings- Type:
Block
- Description:
Details on filter used for electronic couplings in the output
MaxEnergy- Type:
Float
- Default value:
10.0
- Unit:
eV
- Description:
Max. energy (in eV) of diabatic states
MaxEnergyDiff- Type:
Float
- Default value:
0.5
- Unit:
eV
- Description:
Max. energy difference (in eV) between diabatic states
MinCoupling- Type:
Float
- Default value:
0.1
- Description:
Min. coupling value (in meV) that is printed
MinEnergy- Type:
Float
- Default value:
0.0
- Unit:
eV
- Description:
Min. energy (in eV) of diabatic states
FullRun- Type:
Bool
- Default value:
No
- Description:
Include run without restriction of localization of occupied and/or virtual orbitals.
LocalCouplingsOnly- Type:
Bool
- Default value:
No
- Description:
Only account for couplings between local diabatic states
Localize- Type:
Multiple Choice
- Default value:
OccupiedOnly
- Options:
[OccupiedOnly, OccupiedAndVirtual]
- Description:
Localize OccupiedAndVirtual means that separately purely localized excitations and purely charge-transfer excitations are calculated. Localize OccupiedOnly means that an excitation may have local and charge-transfer character, but the excitation only has contributions from occupied orbitals on one fragment. Only relevant in case block key FOCDFT is used or ROSE orbitals are used.
Output- Type:
Multiple Choice
- Default value:
AllCouplings
- Options:
[AllCouplings, FilteredCouplings, AllAndFilteredCouplings]
- Description:
Amount of output
SecondOrder- Type:
Bool
- Default value:
No
- Description:
Include 2nd-order correction to electronic couplings
UseRose- Type:
Bool
- Default value:
No
- Description:
Use ROSE.
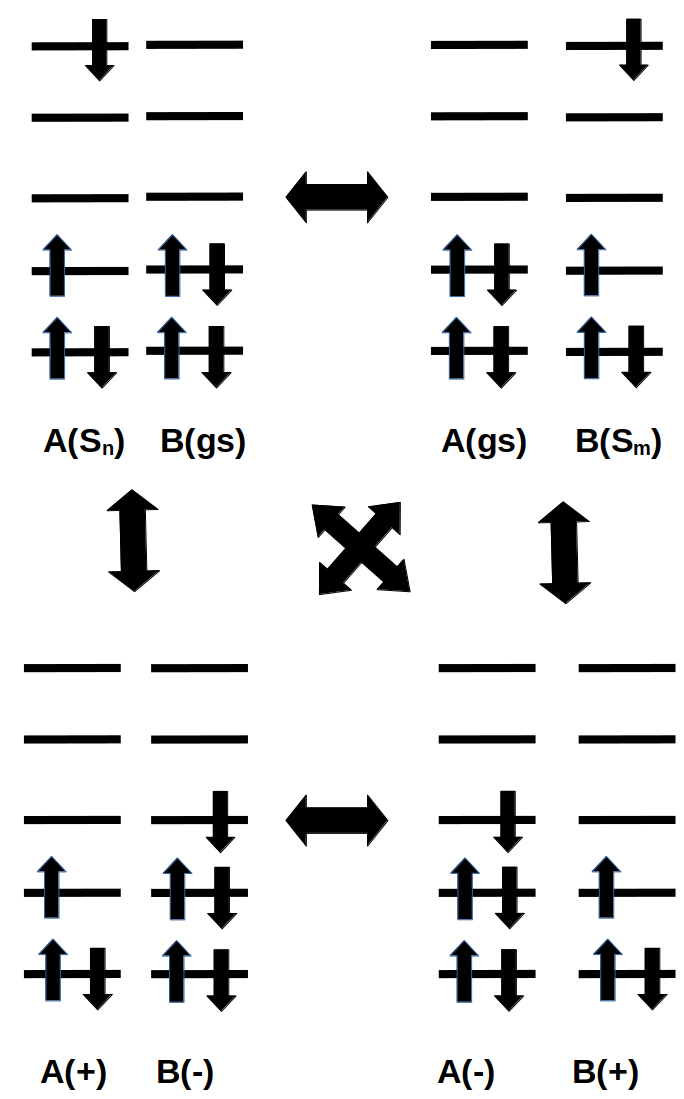
Example electron configuration for charge or exciton hopping from molecule A to molecule B or back. The hopping can occur from the diabatic state where A is in an excited state and B is in the ground state to the diabatic state where A is in the ground state and B is in an excited state or to a diabatic state where both A and B are charged (CT state).
With FOCDFT oscillator strengths can be calculated for the absorption of light to localized diabatic states. Thus with FOCDFT it is possible to investigate, for example, the absorption process where A is locally excited, which is followed by a charge-separation process (like in photosynthesis):
One could also study a reverse process (like in OLED devices):
In order to calculate couplings from LEs from one fragment and CT excitations coming from the same fragment, it is best to set the subkey ExcitonTransfer%Localize to OccupiedAndVirtual.
References