Nudged Elastic Band (NEB)¶
The Nudged Elastic Band (NEB) method [1] can be used to find a reaction path and the transition state between a reactant and a product state.
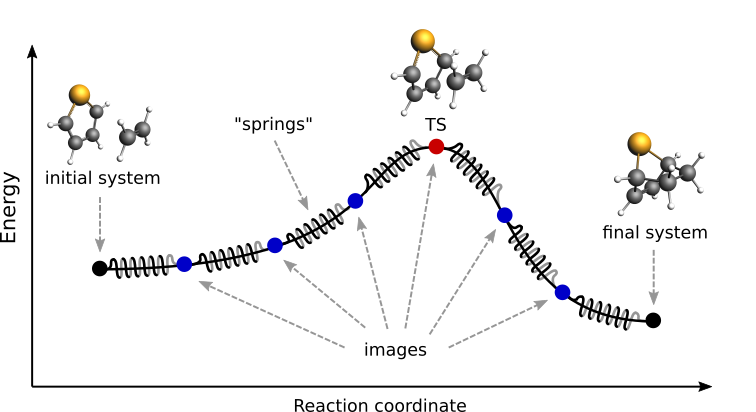
Fig. 1 Pictorial representation of a reaction path computed with NEB¶
At the beginning of a NEB calculation, the geometry of the initial and final systems are optimized to minimize their energy (unless the OptimizeEnds option is set to False).
Then, a rough approximation of the reaction path is build: a set of images is created by performing a linear interpolation between the initial and final systems. If there is more than two systems provided, a spline interpolation is performed between the initial, all the intermediate (in the lexicographical order) and the final system (see below).
Finally, a reaction path is found by performing a simultaneous optimization of all the images. In the NEB method the images are not independent from each other. The force on each image depend on its neighboring images: at each step the forces parallel to the reaction path are eliminated and a so-called spring force is added that tries to keep each image in the middle between its neighbors. This does not let images slide to the initial or final reaction state and ensures that they are evenly distributed along the reaction path.
During the NEB path optimization, a climbing image algorithm is used to drive the highest-energy image in the path to the transition state (unless the Climbing option is set to False).
Be aware that NEB is a computationally expensive method, typically involving hundreds if not thousands of energy and gradients evaluations.
Input¶
A NEB calculation in AMS is triggered by setting the Task to NEB:
Task NEB
The NEB method requires two or more input systems. The first, unnamed system is used as the initial system and the system called final is used as a final system. These two systems are mandatory, unless the LoadPath feature is used, see below. This is an example of system-definitions for a HCN isomerization reaction:
Task NEB
# This is the initial system:
System
Atoms
C 0.0000 0.0000 0.0000
N 1.1800 0.0000 0.0000
H 2.1960 0.0000 0.0000
End
End
# This is the final system (note the header 'final' in the next line):
System final
Atoms
C 0.0000 0.0000 0.0000
N 1.1630 0.0000 0.0000
H -1.0780 0.0000 0.0000
End
End
Optionally, more systems can be used to provide a better approximation for the reaction path. An intermediate system can have any name, but the name determines its position with respect to others. The intermediate systems are ordered lexicographically using their (case-sensitive) names and then placed evenly in that order between the initial and the final states. After that, the spline interpolation between the geometries (including lattice parameters) will be used to generate the initial images. For example, if you call the intermediate systems a, b, and c then the initial NEB path will be constructed by interpolating along the initial-a-b-c-final chain.
When providing more than two input systems it may be a good idea to optimize the end points in advance and set OptimizeEnds to False to prevent creating an unbalanced reaction path. The reason is that the interpolation is performed after the end points are optimized.
For molecular systems, the interpolation can be done either in the redundant internal or in the Cartesian coordinates. For periodic systems, the interpolation can only be done in the Cartesian coordinates.
When using only two systems (the initial and the final) for linear interpolation, AMS will may optionally apply a slightly modified version of the image dependent pair potential (IDPP) method described in [2], see PreOptimizeWithIDPP below. The differences with the original method are: (a) the initial structure of an image is obtained using interpolation in the redundant internal or Cartesian coordinates as described above, and (b) the weighting factor is using the target distance instead of the current one, which avoids putting too much weight on very short distances.
Note that not only the atomic coordinates, but also the lattice parameters and the charge (if non-zero) must be set for all input systems.
Important
The order in which atoms are specified in the System%Atoms blocks must be the same for all systems. The order of the atoms must be consistent because the images-interpolation algorithm maps the n-th atom of the initial system to the n-th atom of the final system.
If you have run a PESScan calculation for the system and would like to do NEB, or would like to repeat NEB for it with a different number of images then you can use the LoadPath feature. You need to provide the file name from which to load the path using the File key. The program will load the last geometries for the path points found in the file and use them for interpolation. Note that this will replace both the initial and the final point. Neither the initial nor the final geometry needs to be present in the input in this case because, if present, they will be replaced.
You can choose to use only part of the path, for example if the the PESScan revealed more than one barrier or if its end points do not correspond to minima on the path. In such a case, a list of point to be included in the interpolation should be specified using the Points key. Remember that you can specify a list of integers as i:j, for example to include points 13 through 25 use Points 13:25.
Alternatively, you can choose to use arbitrary geometries from the History section of the ams.rkf file. In this case you can specify their indices in the Geometries key. When using the Geometries key, the ams.rkf does not have to come from a PESScan or an NEB calculation. It just needs to have the History section with some geometries. It may come, for example, from molecular dynamics.
NEBLoadPath- Type:
Block
- Description:
Provide details about the trajectory to get the initial NEB path from. PESScan and NEB trajectories are supported. Only the last geometry for each point on the trajectory is considered.
File- Type:
String
- GUI name:
Initial path file
- Description:
Provide an ams.rkf file to load the initial path from. All geometries of this calculation, including initial and final, will be taken from the History section of the file. Note that for a PESScan it should be a 1D path.
Geometries- Type:
Integer List
- GUI name:
Raw geometry indices
- Description:
Raw indices of the geometries from the History section. By default the last geometry of each path point is used.
Points- Type:
Integer List
- GUI name:
Path points
- Description:
By default the whole path is used, which may sometimes be not desirable. For example when a PESScan revealed multiple barriers. In this case one can specify indices of the path points to be used. The last geometry of the specified path point will be loaded.
All NEB-specific options are specified in the NEB input block:
NEB
Climbing Yes/No
ClimbingThreshold float
Images integer
InterpolateInternal Yes/No
InterpolateShortest Yes/No
Iterations integer
Jacobian float
LoadPath
File string
Geometries integer_list
Points integer_list
End
MapAtomsToCell Yes/No
OldTangent Yes/No
OptimizeEnds Yes/No
OptimizeLattice Yes/No
Parallel
nCoresPerGroup integer
nGroups integer
nNodesPerGroup integer
End
PreOptimizeWithIDPP Yes/No
ReOptimizeEnds Yes/No
Restart string
Skewness float
Spring float
End
All keys of the NEB block have reasonable defaults or are optional. Thus, in principle, the NEB block can be omitted altogether. These are the main options:
NEBImages- Type:
Integer
- Default value:
8
- GUI name:
Number of images
- Description:
Number of NEB images (not counting the chain ends). Using more images will result in a smoother reaction path and can help with convergence problems, but it will also increase the computation time.
Iterations- Type:
Integer
- GUI name:
Maximum number of iterations
- Description:
Maximum number of NEB iterations. The default value depends on the number of degrees of freedom (number of images, atoms, periodic dimensions).
Spring- Type:
Float
- Default value:
1.0
- Unit:
Hartree/Bohr^2
- GUI name:
Spring value
- Description:
Spring force constant in atomic units.
Skewness- Type:
Float
- Default value:
1.0
- GUI name:
Skewness
- Description:
Degree of how much images are shifted towards or away from the TS, which may help tackle problems with a long reaction path (for example involving a loose adsorption complex) without needing too many images. A value greater than 1 will make sure that images are concentrated near the transition state. The optimal value depends on the path length, the number of images (larger [Skewness] may be needed for a longer path and fewer images). Technically [Skewness] is equal to the ratio between the optimized distances to the lower and the higher neighbor image on the path.
Climbing- Type:
Bool
- Default value:
Yes
- GUI name:
Climb highest image to TS
- Description:
Use the climbing image algorithm to drive the highest image to the transition state.
ClimbingThreshold- Type:
Float
- Default value:
0.0
- Unit:
Hartree/Bohr
- GUI name:
CI force threshold
- Description:
Climbing image force threshold. If ClimbingThreshold > 0 and the max perpendicular force component is above the threshold then no climbing is performed at this step. This entry can be used to get a better approximation for the reaction path before starting the search for the transition state. A typical value is 0.01 Hartree/Bohr.
InterpolateInternal- Type:
Bool
- Default value:
Yes
- GUI name:
Interpolate in Internal coordinates
- Description:
The initial NEB image geometries are calculated by interpolating between the initial and the final state. By default, for non-periodic systems the interpolation is performed in internal coordinates but the user can choose to do it in the Cartesian ones. For periodic systems the interpolation is always done in Cartesian coordinates. If PreOptimizeWithIDPP is set then the path may be further refined using the image-dependent pair potential (IDPP).
InterpolateShortest- Type:
Bool
- Default value:
Yes
- GUI name:
Interpolate across cell boundary
- Description:
Allow interpolation across periodic cell boundaries. Set to false if an atom is intended to move more than half across the simulation box during reaction.
PreOptimizeWithIDPP- Type:
Bool
- Default value:
No
- GUI name:
Use IDPP
- Description:
(Experimental) When there is only initial and final system available, the image-dependent pair potential (IDPP, doi: 10.1063/1.4878664) can be used to determine the initial NEB path by interpolating all interatomic distances between the two points and optimizing intermediate images towards them. The optimization starts from the geometries obtained using the selected interpolation options.
OptimizeEnds- Type:
Bool
- Default value:
Yes
- GUI name:
Optimize reactants/products
- Description:
Start the NEB with optimization of the reactant and product geometries.
Restart- Type:
String
- GUI name:
Restart from
- Description:
Provide an ams.rkf file from a previous NEB calculation to restart from. It can be an unfinished NEB calculation or one performed with different engine parameters.
ReOptimizeEnds- Type:
Bool
- Default value:
No
- GUI name:
Re-optimize reactants/products
- Description:
Re-optimize reactant and product geometries upon restart.
The following keys are related to solid-state NEB (SS-NEB):
NEBOptimizeLattice- Type:
Bool
- Default value:
No
- GUI name:
Optimize lattice
- Description:
Turn on the solid-state NEB (SS-NEB).
Jacobian- Type:
Float
- GUI name:
Jacobian value
- Description:
Scaling factor used to convert the lattice strain to a NEB coordinate value. Default value: sqrt(N)*(V/N)^(1/d), where V - lattice volume (area for 2D, length for 1D), N - number of atoms, and d - number of periodic dimensions.
MapAtomsToCell- Type:
Bool
- Default value:
Yes
- GUI name:
Map atoms to cell
- Description:
Translate atoms to the [-0.5,0.5] cell before every step. This option cannot be disabled for SS-NEB.
At each iteration, the images may be computed in parallel. The parallel execution is normally configured completely automatically, but users can override the automatic parallelization using the keys in the Parallel block.
NEBParallel- Type:
Block
- Description:
Options for double parallelization, which allows to split the available processor cores into groups working through all the available tasks in parallel, resulting in a better parallel performance. The keys in this block determine how to split the available processor cores into groups working in parallel.
nCoresPerGroup- Type:
Integer
- GUI name:
Cores per group
- Description:
Number of cores in each working group.
nGroups- Type:
Integer
- GUI name:
Number of groups
- Description:
Total number of processor groups. This is the number of tasks that will be executed in parallel.
nNodesPerGroup- Type:
Integer
- GUI name:
Nodes per group
- Description:
Number of nodes in each group. This option should only be used on homogeneous compute clusters, where all used compute nodes have the same number of processor cores.
The following keys modify other aspects of the NEB and should, in principle, be left to their defaults:
NEBOldTangent- Type:
Bool
- Default value:
No
- GUI name:
Use old tangent
- Description:
Turn on the old central difference tangent.
Frozen atom constraints¶
It is possible to perform NEB with part of the system frozen, using any of the following keys of the Constraints block or a combination thereof:
ConstraintsAtom- Type:
Integer
- Recurring:
True
- Description:
Fix the position of an atom. Just one integer referring to the index of the atom in the [System%Atoms] block.
AtomList- Type:
Integer List
- Recurring:
True
- Description:
Fix positions of the specified atoms. A list of integers referring to indices of atoms in the [System%Atoms] block.
FixedRegion- Type:
String
- Recurring:
True
- Description:
Fix positions of all atoms in a region.
Note: the frozen atom constraints will be enforced both during the geometry optimizations of the initial and final systems and during the NEB optimization.
Optimizations and convergence criteria¶
The NEB path is optimized using a limited-memory BFGS (l-BFGS) method where the system being optimized is a union of all NEB images with their respective molecular and spring forces.
The NEB convergence thresholds are defined in the GeometryOptimization%Convergence block. NEB is considered converged when the following criteria are satisfied:
the change in the highest image energy must be less than [GeometryOptimization%Convergence%Energy]
the max atomic force component for the highest image must be less than [GeometryOptimization%Convergence%Gradients]
the max atomic force component for all other images must be less than ten times the [GeometryOptimization%Convergence%Gradients] value.
If the optimization of the initial NEB end point fails to converge, you can try using the FIRE optimization method.
Output¶
Results are printed to the text output and stored in the binary result file ‘ams.rkf’. In the ‘ams.rkf’ file, NEB calculation results are stored in the History section just like in a normal geometry optimization. The NEB section of the RKF file contains additional, NEB-specific, information.
The NEB reaction path can be visualized using the AMSmovie GUI module.
Troubleshooting¶
In case the geometry optimization of the initial and final systems fails: try using the FIRE optimization method
In case the optimization of the NEB path does not converge:
make sure that the order in which the atoms are defined is consistent between the initial and final systems (see the important note in the NEB input section);
try increasing the number of NEB images;
try tweaking the TrustRadius or TrialStep options (see Limited-memory BFGS) ;
try specifying one or more intermediate systems.
References