Screening for cocrystals¶
In this section, we provide an example application that can be used as a template for many high-throughput screening scripts. Cocrystals are crystals formed from two or more compounds in a defined stoichiometry. There are many uses for cocrystals, especially for pharmaceutical applications where one compound is an active pharmaceutical ingredient (API). This example problem screens multiple compounds for their potential as components of a (1:2) cocrystal with Itraconazole. This problem uses the excess enthalpy for a hypothetical supercooled liquid phase as a proxy for cocrystallization affinity. The rankings of the solvents are in good agreement with model and experimental results for this problem given in [1] .
Download relevant coskf file: coskf_Hex.zip
Python code using pyCRS¶
[show/hide code]
from scm.plams import Settings, init, finish, CRSJob, config, JobRunner, KFFile
from pyCRS.Database import COSKFDatabase
from pyCRS.CRSManager import CRSSystem
from pathlib import Path
import matplotlib.pyplot as plt
import numpy as np
init()
####### Step 1 : add compounds to a pyCRS.databse. Ensure coskf_Hex is downloaded #######
####### Note: Ensure to download the coskf_Hex before running the script ##############
db = COSKFDatabase("my_coskf_db.db")
coskf_path = Path("./coskf_Hex")
if not coskf_path.exists():
raise OSError(f"The provided path does not exist. Exiting.")
CAS_ref = { "tartaric_acid" : "526-83-0",
"fumaric_acid" : "110-17-8",
"succinic_acid" : "110-15-6",
"malic_acid" : "6915-15-7",
"glutaric_acid" : "110-94-1",
"malonic_acid" : "141-82-2",
"adipic_acid" : "124-04-9",
"maleic_acid" : "110-16-7",
"itz" : "84625-61-6"}
for file in coskf_path.glob("*.coskf"):
for key, val in CAS_ref.items():
if(key in file.name):
CAS = val
name = key.replace("_"," ")
if(name == "itz"): name = "itraconazole"
db.add_compound(file.name,cas=CAS,name=name,coskf_path=coskf_path)
####### Step2 : Iterately set up calculation for solubility and activitycoef #######
## Set up for parallel run ##
if True:
config.default_jobrunner = JobRunner(
parallel=True, maxjobs=8
) # Set jobrunner to be parallel and specify the numbers of jobs run simutaneously (eg. multiprocessing.cpu_count())
config.default_jobmanager.settings.hashing = None # Disable rerun prevention
config.job.runscript.nproc = 1 # Number of cores for each job
config.log.stdout = 0 # suppress plams output default=3
solv_name = [x.replace("_"," ") for x in list(CAS_ref.keys())[0:-1]]
System = CRSSystem()
System2 = CRSSystem()
for co_solvent in solv_name:
mixture = {}
mixture[co_solvent] = 0.33333
mixture["itraconazole"] = 0.66667
System.add_Mixture(
mixture, database="my_coskf_db.db", temperature=298.15, problem_type="VAPORPRESSURE", conformer=True, jobname="conf"
)
System2.add_Mixture(
mixture, database="my_coskf_db.db", temperature=298.15, problem_type="VAPORPRESSURE", conformer=False, jobname="Emin"
)
System.runCRSJob()
System2.runCRSJob()
####### Step3 : Output processing to retrive results for plotting #######
print("Solvent".ljust(14), "Hex_Emin", "Hex_conf", "Population of solvent's conformers")
Hex_conf = []
Hex_Emin = []
for out, out2, name in zip(System.outputs, System2.outputs, solv_name):
res = out.get_results()
res2 = out2.get_results()
Hex = res["excess H"]
Hex2 = res2["excess H"]
Hex_conf.append(Hex)
Hex_Emin.append(Hex2)
compositions = out.get_multispecies_dist()[0]
print(name.ljust(15), end="")
print(f"{Hex2:.3f}", end=" ")
print(f"{Hex:.3f}", end=" ")
for conf, frac in compositions.items():
print(f"{frac[0]:.5f}", end=" ")
print("")
if True:
plt_index = [i for i in range(len(Hex_conf))]
plt.xlabel("Excess enthalpy (kcal/mol)")
plt.barh(plt_index, Hex_conf, zorder=3)
plt.yticks(plt_index, solv_name)
plt.grid(axis="x", ls="--", zorder=0)
plt.gca().invert_xaxis()
# plt.savefig('./Cocrystal_screening.png',dpi=300)
plt.title("Hex with conformers")
plt.tight_layout()
plt.show()
if True:
plt_index = [i for i in range(len(Hex_Emin))]
plt.xlabel("Excess enthalpy (kcal/mol)")
plt.barh(plt_index, Hex_Emin, zorder=3)
plt.yticks(plt_index, solv_name)
plt.grid(axis="x", ls="--", zorder=0)
plt.gca().invert_xaxis()
# plt.savefig('./Cocrystal_screening_Emin.png',dpi=300)
plt.title("Hex with Lowest energy conformer")
plt.tight_layout()
plt.show()
Python code using PLAMS¶
[show/hide code]
import os, time
import multiprocessing
import numpy as np
import matplotlib.pyplot as plt
from scm.plams import Settings, init, finish, CRSJob, config, JobRunner
def set_CRSJob_Hex_conf(index, ncomp, coskf, database_path, cal_type, method, temperature, comp_input={}, comp_type=[]):
s = Settings() # initialize a settings object
s.input.property._h = cal_type # specify problem type
s.input.method = method # specify method
s.input.temperature = temperature # specify temperature
compounds = [Settings() for i in range(ncomp)] # initialization of compounds
for i in range(ncomp):
if len(comp_type) > i:
if "conf" in comp_type[i]:
form = [Settings() for i in range(len(coskf[i]))] # initialize compound in multiple form
for j in range(len(coskf[i])):
form[j]._h = os.path.join(
database_path, coskf[i][j]
) # specify conformer's coskf file for each form
compounds[i].form = form
else:
compounds[i]._h = os.path.join(database_path, coskf[i]) # specify absolute directory of coskf file
else:
compounds[i]._h = os.path.join(database_path, coskf[i]) # specify absolute directory of coskf file
for column, value in comp_input.items(): # specify compound's information through comp_input, for example
if value[i] != None: # column: frac1, meltingpoint, hfusion
compounds[i][column] = value[i]
s.input.compound = compounds
my_job = CRSJob(settings=s) # create jobs
my_job.name = cal_type + "_" + str(index) # specify job name
return my_job
init()
Parallel_run = True
Plot_option = True
if Parallel_run:
config.default_jobrunner = JobRunner(
parallel=True, maxjobs=8
) # Set jobrunner to be parallel and specify the numbers of jobs run simutaneously (eg. multiprocessing.cpu_count())
config.default_jobmanager.settings.hashing = None # Disable rerun prevention
config.job.runscript.nproc = 1 # Number of cores for each job
config.log.stdout = 1 # suppress plams output default=3
###----INPUT YOUR DATA HERE----###
database_path = os.path.join(os.getcwd(), "coskf_Hex")
cal_type = "VAPORPRESSURE"
method = "COSMORS"
ncomp = 2
solute = "itz_c1.coskf"
# a list of different conformers for the screening each line contains 3 conformers of the same molecule
solvents = [
["tartaric_acid_c1.coskf", "tartaric_acid_c2.coskf", "tartaric_acid_c3.coskf"],
["fumaric_acid_c1.coskf", "fumaric_acid_c2.coskf", "fumaric_acid_c3.coskf"],
["succinic_acid_c1.coskf", "succinic_acid_c2.coskf", "succinic_acid_c3.coskf"],
["malic_acid_c1.coskf", "malic_acid_c2.coskf", "malic_acid_c3.coskf"],
["glutaric_acid_c1.coskf", "glutaric_acid_c2.coskf", "glutaric_acid_c3.coskf"],
["malonic_acid_c1.coskf", "malonic_acid_c2.coskf", "malonic_acid_c3.coskf"],
["adipic_acid_c1.coskf", "adipic_acid_c2.coskf", "adipic_acid_c3.coskf"],
["maleic_acid_c1.coskf", "maleic_acid_c2.coskf", "maleic_acid_c3.coskf"],
]
temperature = 298.15 # K
# store input data along with the necessary thermal property in comp_input dictionary
comp_input = {}
comp_input["frac1"] = [0.33333, 0.66667] # the stoichiometric ratio of the co-crystal (solvent:solute)
# enter 'conf' for compound with multiple conformers; '' for compound with single structure
comp_type = ["conf", ""]
###----INPUT END----###
outputs = []
solv_name = []
for index, solv in enumerate(solvents):
solv_name.append(solv[0].replace("_c1.coskf", ""))
coskf = [solv, solute]
comp_input["name"] = [solv[0].replace("_c1.coskf", ""), solute.replace("_c1.coskf", "")]
job = set_CRSJob_Hex_conf(index, ncomp, coskf, database_path, cal_type, method, temperature, comp_input, comp_type)
outputs.append(job.run())
finish()
# In a parallel run, the get_results function will wait for the completion of the corresponding jobs.
results = []
excess_h = []
print("")
print("Solvent".ljust(14), "Population of solvent's conformers")
for out, name in zip(outputs, solv_name):
res = out.get_results()
results.append(res)
excess_h.append(res["excess H"])
compositions = out.get_multispecies_dist()[0]
print(name.ljust(15), end="")
for conf, frac in compositions.items():
print(f"{frac[0]:.5f}", end=" ")
print("")
print("")
print("Solvent".ljust(15), "Excess enthalpy (kcal/mol)")
for (
name,
Hex,
) in zip(solv_name, excess_h):
print(name.ljust(15), round(Hex, 5))
if Plot_option:
plt_index = [i for i in range(len(excess_h))]
plt.xlabel("Excess enthalpy (kcal/mol)")
plt.barh(plt_index, excess_h, zorder=3)
plt.yticks(plt_index, solv_name)
plt.grid(axis="x", ls="--", zorder=0)
plt.gca().invert_xaxis()
plt.tight_layout()
# plt.savefig('./Cocrystal_screening.png',dpi=300)
plt.show()
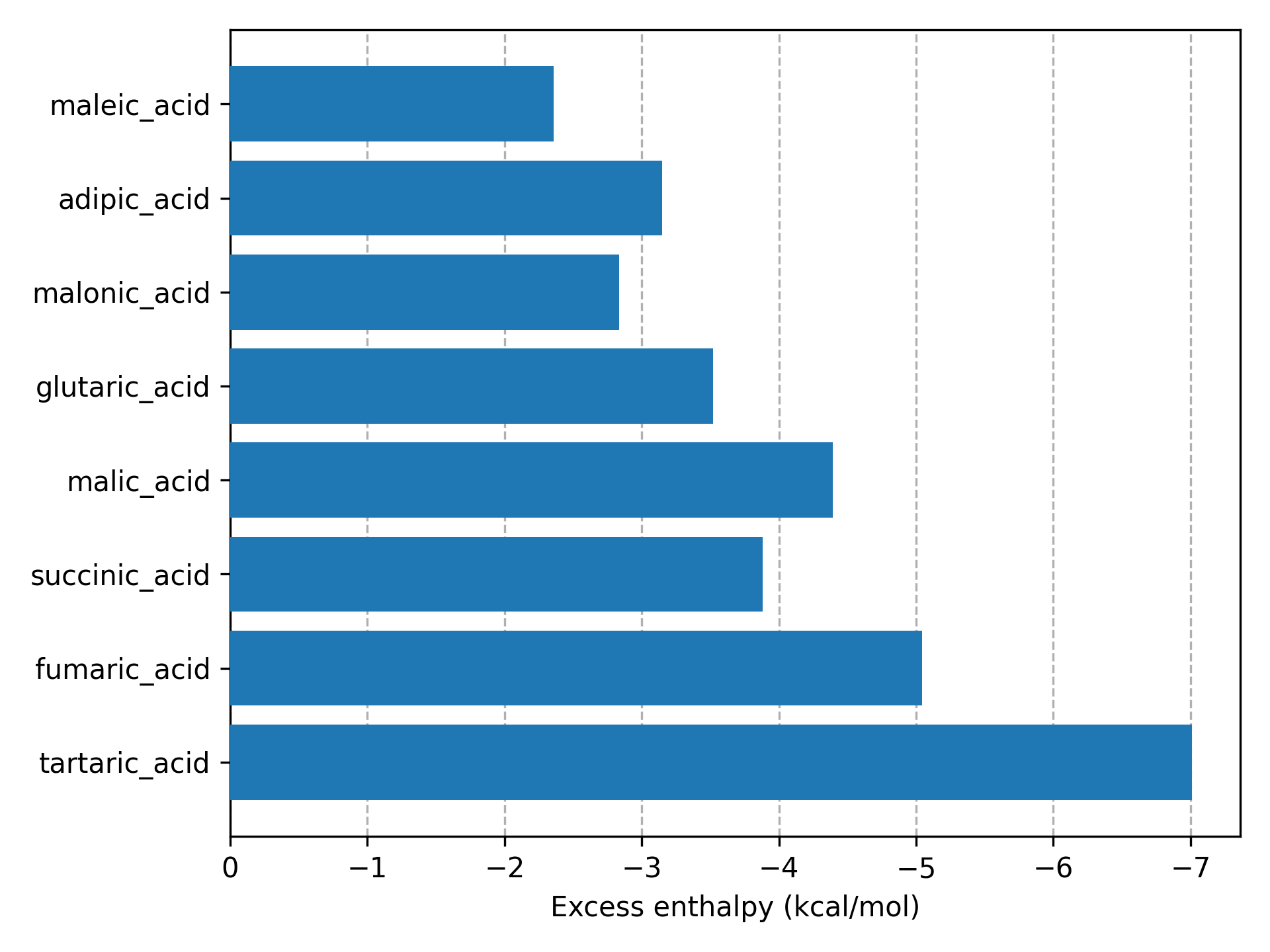
This figure (produced by the code) shows the excess enthalpy values of all solvents in a supercooled liquid mixture with Itraconazole. The lowest 4 excess enthalpy values correspond to 4 solvent for which a stable co-crystal with Itraconazole is known [1] .