ChemTraYzer2¶
ChemTraYzer2 (CT2) is a tool for post-processing reactive molecular dynamics (MD) trajectories. The purpose of CT2 is to detect and distinguish the reactive events that occur, construct a database of unique reactions from these events, and then calculate aggregate kinetic and population properties for the trajectory. Practically speaking, CT2 is capable of greatly simplifying MD simulations into a set of useful values such as reaction rate constants [1], net fluxes for all chemical species, and occurrence counts for all reactions. ChemTraYzer2 is the successor of ChemTraYzer.
See also
The GUI tutorial Detecting reactions with ChemTraYzer 2: Hydrogen combustion with ReaxFF will show you how to set up and perform a ChemTraYzer2 analysis using the Graphical User Interface.
New in ChemTraYzer2-2023¶
trajectory population analysis
support for trajectories with a non-constant number of atoms
an improved reaction rates calculator
additional output files for population statistics
Important information for using ChemTraYzer2¶
Bond orders are necessary for post-processing MD trajectories with ChemTraYzer2. CT2 does not estimate bond orders but instead uses those computed by the MD engine used to run the simulation. Though most AMS engines can compute bond orders, there are some that cannot (see Summary of engine capabilities). CT2 can still be used with these engines, but a bond guessing algorithm must be used to estimate the bond orders. This can be done by specifying the following settings in the MD input.
Important
When preparing MD simulations for use with CT2, it is recommended to set the BondOrders variable in the Properties block to Yes. This will ensure that bond orders are calculated and stored. Depending on the chosen engine’s capabilities, either it will supply bond orders or a bond guessing algorithm will be used. More information on this setting can be found in here
The quality of the ChemTraYzer2’s analysis depends partially on the quality of the bond orders provided, but it is more dependent on the connectivity information (i.e., whether or not there is a bond between two atoms).
The ChemTraYzer2 algorithm¶
The following is a summary of the steps taken by ChemTraYzer2 while post-processing a MD trajectory. All of these steps are automatically conducted by ChemTraYzer2, so it is not necessary to understand them in detail in order to use ChemTraYzer2. This section is simply intended to provide the interested user with more technical information about the algorithm.
(1) Identifying all bond breaking and bond forming events in the the MD trajectory
Bond changes are fundamental to chemical reactions, and the first step of ChemTraYzer2 is to analyze the MD trajectory and detect all bond change events that occur. ChemTrayzer2 defines a bond change event as either of the following:
Bond formation – this occurs when the bond order between 2 atoms crosses the
BondFormationThresholdparameter between 2 MD frames. More specifically, this means the bond order between 2 atoms must be below theBondFormationThresholdin one frame and then above it in the subsequent frame.Bond breakage – this occurs when the bond order between 2 atoms decreases to below the
BondBreakingThreshold. It is defined analogously to bond formation.
(2) Filtering and combining all bond change events into stable reactions using the TStable criterion
Many bond change events in a MD trajectory might represent the formation of short-lived intermediates that do not need to be explicitly included in the complete reaction. These intermediates, though perhaps important to the mechanism, do not affect the overall reactants and products of a reaction and may introduce unwanted complexity to ChemTraYzer2 output. For this reason, the adjustable parameter TStable is used to filter out reactive intermediates which exist for an amount of time less than TStable. An example of using TStable to filter reactions is provided below.

Fig. 1 An example of how ChemTraYzer2 filters reaction events based on the TStable criterion. In this reaction network, all species in red are determined to be short-lived intermediates and do not appear in the final reactions.¶
(3) Removing all reactions that have the same reactants and products
It is not uncommon for chemical equilibria to be observed in certain MD trajectories. Certain equilibria occur on a very short time-scale, meaning a series of bond change events may be filtered out using the TStable criterion. In these cases, the remaining reaction can have identical molecules on both sides of the reaction, as shown below.
These reactions are removed from the final reaction list as they have no effect on net species fluxes, rate constants, etc.
Note
Reactions that involve bond changes but result in the same molecules will also be filtered. For example, the following proton transfer will not be included in the final reaction list: \(\mathrm{H_3O^+} + \mathrm{H_2O} \rightarrow \mathrm{H_2O} + \mathrm{H_3O^+}\). Options for including these reactions will be present in the next version of CT2.
(4) Aggregating equivalent reactions
After the filtering steps are complete, all equivalent reaction events are combined into a set of unique reactions that have occurred in the MD trajectory. More specifically, the reaction event \(A\rightarrow B\) may have happened multiple times in the trajectory, and each of these will count toward one occurrence of the \(A \rightarrow B\) reaction. More detail about determining when two reactions (or molecules) are equivalent is provided in the following section.
Distinguishing reactions with ChemTraYzer2¶
In ChemTraYzer2, reactions are determined to be equivalent using a very straightforward condition: two reactions (R1 and R2) are equivalent if the sets of reactant/product molecules of R1 and the sets of reactant/product molecules of R2 are equivalent. Comparing reactions in this way requires defining the equivalence of two individual molecules, and this is more challenging to assess. In the original ChemTraYzer, molecule equivalence is determined via a comparison of canonical SMILES strings. Though SMILES can represent a large number of chemical structures, they fall short in representing the complete space of chemical reactions. For this reason, ChemTraYzer2 evaluates each molecule using a subgraph-based descriptor, which is generalizable to the complete reactive chemical space. ChemTraYzer2’s subgraph descriptor builds local atomic environments using a breath-first search of each atom in a molecule, evaluates a unique hash value for each atom, and finally sums these hash values to produce a unique hash value for each unique molecule. This is summarized in the figure below.
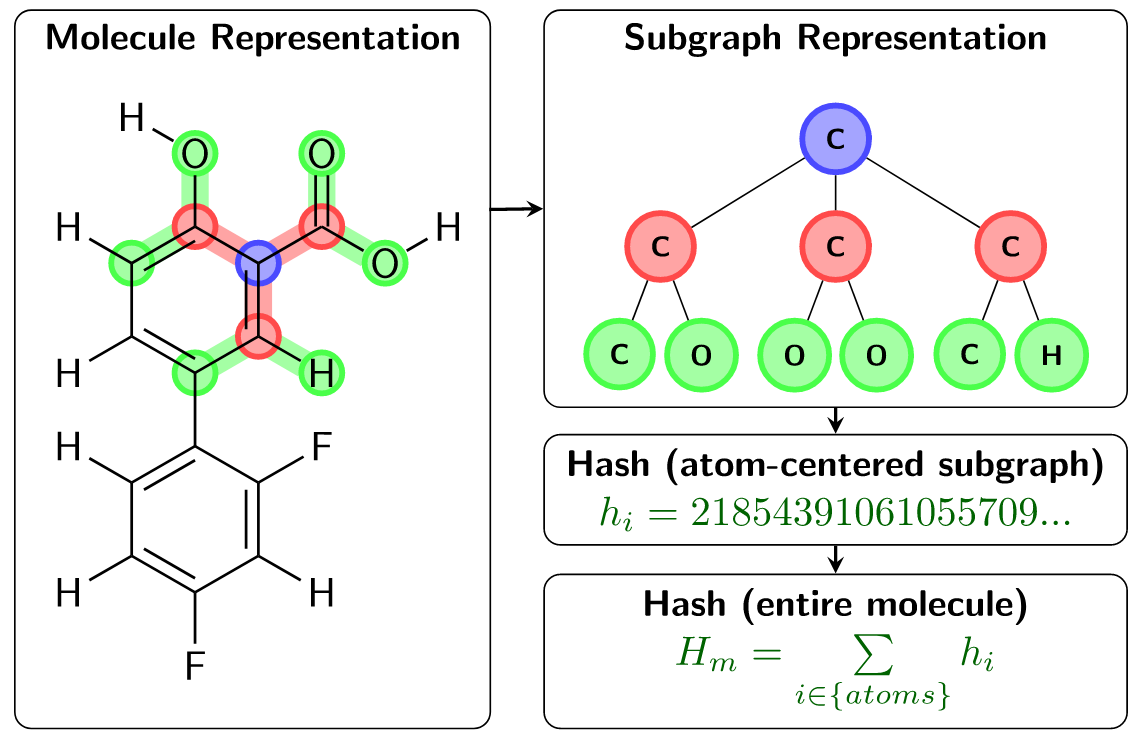
Fig. 2 The subgraph-based descriptors used to distinguish molecules in ChemTraYzer2¶
Note
The current version of the subgraph descriptors do not distinguish stereoisomers
Using ChemTraYzer2 from the GUI¶
ChemTraYzer2 is fully supported in the AMS GUI. A thorough description of using the GUI can be found in the ChemTraYzer2 GUI tutorial.
Tips for getting the most out of ChemTraYzer2¶
The MD simulation¶
It is important to ensure the simulation is on a time scale that is long enough to observe multiple reaction events. Multiple occurrences of reactions improve the accuracy of calculations of kinetic parameters such as reaction rate constants.
The sampling frequency of MD trajectories should be sufficiently small to observe all important reactions. In AMS MD simulations, this is controlled by the
SamplingFreqkeyword in theTrajectoryblock (see the Molecular dynamics page for more details). If the sampling frequency is too large, important reaction events may not be detected by ChemTraYzer2, which will have an effect on the quality of the reported properties. A rough recommendation would be to set the sampling frequency to at most 10 for a time step of 0.25 fs, but the best value for this parameter depends on the temperature of the simulation.
ChemTraYzer2 Settings¶
Set the
TStableparameter to an appropriate value. Typically, the default value will work for many applications. However, the user can adjust this parameter to generate output on the spectrum between many reactive intermediates (lowTStable) and a summary of only the main reactions (highTStable). Generally, it is best to adjustTStableto a level where all important intermediates are long-lived enough to appear in the final output. You may want to perform a few CT2 analysis using different values forTStableto see how this affects the results.Set the
BondBreakingThresholdandBondFormationThresholdparameters to appropriate values for the chemical system. The default values are suitable for most types of systems, but these threshold values may need to be changed in certain cases (e.g., the MD engine calculates bond orders with a systematic error, bonds in the system have partial ionic character, etc.).Set the rate confidence interval
RateConfidenceto adjust bounds for the reaction rate constants. CT2 assumes the number of observed reactive events are distributed according to a Poisson distribution, where the expected value is used to calculate the reaction rate constant. The confidence interval specifies what ratio of the event counts will fall between the lower and upper bounds, with the condition that both bounds represent an equal number of events. Usually, a confidence interval of 95% is used, which corresponds roughly to \(2\sigma\) in a normal distribution. For more details about this approach, see [1].
Minimal input¶
This is the minimal input script for performing a chemtrayzer2 analysis of your MD trajectory:
#!/bin/sh
$AMSBIN/chemtrayzer2 << EOF
Trajectory
Path path/to/the/ams/results/folder
End
EOF
Input options¶
Several input options can be specified in the chemtrayzer2 input.
The trajectory the user wants to analyze can be specified in the Trajectory block:
Trajectory
FinalFrame integer
FirstFrame integer
Path string
End
Trajectory- Type:
Block
- Description:
Info regarding the trajectory to analyze.
FinalFrame- Type:
Integer
- Default value:
-1
- Description:
Last frame of the trajectory to analyze.
FirstFrame- Type:
Integer
- Default value:
1
- Description:
First frame of the trajectory to analyze.
Path- Type:
String
- Description:
The path to ams results dir of an AMS calculation. This folder must contain a ams.rkf file.
Reaction detection options can be specified in the ReactionDetection block:
ReactionDetection
BondBreakingThreshold float
BondFormationThreshold float
InitialBondThreshold float
TStable float
End
ReactionDetection- Type:
Block
- Description:
Parameters for the the reaction detection algorithm.
BondBreakingThreshold- Type:
Float
- Default value:
0.3
- Description:
The bond-order threshold for bond breaking. If the bond order of a bond goes below this value, the bond is considered broken.
BondFormationThreshold- Type:
Float
- Default value:
0.8
- Description:
The bond-order threshold for bond formation. If the bond order between two atoms goes above this value, then this will be considered to be a new bond.
InitialBondThreshold- Type:
Float
- Description:
The bond-order threshold for determining the connectivity for the first frame of the simulation. If not specified, the value in BondFormationThreshold will be used instead.
TStable- Type:
Float
- Default value:
10.0
- Unit:
fs
- GUI name:
T stable
- Description:
The minimum time for a molecule to be considered stable.
Options for the analysis of the reactions:
Analysis
PerformAnalysis Yes/No
RateConfidence float
End
Analysis- Type:
Block
- Description:
Statistical post-detection analysis, includes reaction coefficients calculation.
PerformAnalysis- Type:
Bool
- Default value:
Yes
- Description:
Determine the reaction rate coefficients and statistical errors for the detected reactions.
RateConfidence- Type:
Float
- Default value:
0.9
- Description:
Upper and lower bounds to the rate coefficients will be calculated for this confidence (0 < confidence < 1), assuming a Poisson distribution of the number of reactive events. A value of 0.9 means that the kinetics of 90% of events of one reaction can be described by a coefficient between the bounds.
Options for Output file writing:
Output
CreateLegacyOutput Yes/No
ShowReactionGraph Yes/No
WriteEventsPerTime Yes/No
WriteKF Yes/No
WriteMolPopulation Yes/No
WriteReactions Yes/No
WriteXYZFiles Yes/No
End
Output- Type:
Block
- Description:
Settings for program output and output file generation.
CreateLegacyOutput- Type:
Bool
- Default value:
No
- Description:
Whether to save the reactions, species, and rates as ‘reac.reac.tab’, ‘reac.spec.tab’, and ‘reac.rate.tab’ in the same format as ChemTraYzer 1.
ShowReactionGraph- Type:
Bool
- Default value:
No
- Description:
Whether or not to show the reaction graph at the end of the calculation. Requires the python library matplotlib to be installed.
WriteEventsPerTime- Type:
Bool
- Default value:
No
- Description:
Write two .csv files that contain the number of reactions in every frame (reaction_events_per_time.csv) and the number of bond changes in every frame(bond_change_events_per_time.csv)
WriteKF- Type:
Bool
- Default value:
No
- Description:
Whether to write output to KF
WriteMolPopulation- Type:
Bool
- Default value:
No
- Description:
Write two .csv files: (1) mol_statistics.csv, which contains basic population statistics (counts, averages) for each unique species over the entire trajectory; and (2) mol_population.csv, which provides the count of each unique species in every frame.
WriteReactions- Type:
Bool
- Default value:
Yes
- Description:
Write two .csv files that contain information about (1) all unique reactions (reactions.csv); and (2) all individual reaction events (reaction_events.csv).
WriteXYZFiles- Type:
Bool
- Default value:
No
- Description:
Write XYZ files (geometries) for detected species and XYZ movies for detected reactions into a subfolder named ‘xyz’.
Output¶
Summarizing reactions¶
ChemTraYzer2 produces 2 main output files for summarizing reactions, reaction_events.csv and reactions.csv. These 2 files are produced with the option WriteReactions in the Output block.
reaction_events.csv¶
This file contains a list of all bond breaking or bond forming events. These events are complete reactions that occur for some specific set of molecules at some specific point in the trajectory. Various important properties are included in this file, a few of which are listed below.
Initial frame– the MD frame at which the bond change event beganFinal frame– the MD frame at which the bond change event endedReactants/Products– a SMILES-like representation of molecules involved in the reactionReactants atoms indices/Products atoms indices– the atom indices of the molecules involved in the reaction
reactions.csv¶
This file contains aggregate information about all unique reactions that occurred in the trajectory. A few important properties contained in this file are listed below.
Rate constant– the calculated value of the reaction rate constants. Note that the units for the reaction rate depend on the reaction order.Number of events– the number of times this reaction occurred in the trajectoryReaction event indices– the indices of all reactive events that are equivalent to this reaction. The indices correspond to indices in thereaction_events.csvfile.
Reaction frequency¶
The option WriteEventsPerTime in the Output block will produce two files that detail the accumulated number of reactions and reaction events per frame over the entire trajectory.
reaction_events_per_time.csv¶
Frame– the MD frameTime– the simulation time for the frameEventsthe number of reactions that begin in the specified frame
bond_change_events_per_time.csv¶
Frame– the MD frameTime– the simulation time for the frameEventsthe number of bond change events that occur in the specified frame
Molecular population analysis¶
The option WriteMolPopulation in the Output block will produce two files that provide summary statistics for each unique molecule in the trajectory as well as population counts for all frames.
mol_statistics.csv¶
Molecule hash– the hash value used to identify a moleculeSMILES– the SMILES representation of a molecule, should one be availableAverage count– the average number of molecule over the entire trajectoryAverage conc.– the average concentration (in mol/L) of molecule over the entire trajectoryMann-Kendall value"– a value in the range [-1,1] that indicates whether a molecule behaves more like a reactant (with a maximum value of -1) or a product (with a maximum value of +1). Intermediates are expected to have values around 0.
mol_population.csv¶
Frame– the MD frameTime– the simulation time for the frameCountthe number of a particular molecule in a particular frame
Geometry output¶
The option WriteXYZFiles will produce xyz files for each unique molecule and a series of xyz frames for each unique reaction. These files are named according to the molecule and reaction indices and will be placed into a directory called xyz.
Additional output files¶
In addition to the main csv output files, ChemTraYzer2 generates a gml file (reaction_network.gml) containing the full reaction network.
At the moment, we don’t offer any built-in tool for visualizing or manipulating this file.
The savvy user might want to import and analyze the .gml file using the networkx python library or visualize it with third party graph visualization tools.