Cluster Growth: Cobalt Clusters¶
Here we show a biased structure search strategy for investigating the structural evolution and growth pattern of single-atom clusters. The strategy consists of adding one atom to the lowest energy structures of the n-atoms (frozen) and get the binding site with the largest binding energy, which is a good approximation to the (n+1)-cluster global minimum structure.
The strategy is illustrated using the Co.ff ReaxFF force field for the growth mechanism Co8+Co–>Co9.
The tutorial is divided into two parts: In the first part, we obtain the global minimum for the Co8 cluster by using the Basin Hopping subtask. In the second part, we fix the coordinates of the Co8 global minimum, add a Co atom and use the Process Search subtask to obtain all reaction mechanisms involving it, and with them, the corresponding binding sites. The structure associated with the highest binding energy is the best approximation to the Co9 global minimum structure, and the binding-sites lattice will give us insights into the growth mechanism behind it. The gradual population of the binding sites shows the structural evolution and the growth pattern of these kinds of clusters.
Hereafter, we will use the PES exploration task in the AMS driver with the ReaxFF engine. Configure this by following the steps below:
 →
→  .
.Global Optimization of Co8¶
We need an initial guess for the structure of the Co8 cluster. Thus, create a random structure composed of 8 atoms of Co by using the drawing tools of AMSinput. Avoid a planar configuration for the cluster’s atoms; to do so, take some atoms out of that plane individually if needed. The difficulty is that planar configurations usually converge to a transition state during optimization instead of a local minimum. Then, use the Pre-optimize button on the main panel to get the closest local minimum, make sure to pre-optimize the full structure. Make sure that you have a non-flat structure with none of the atoms collapsed. Otherwise try rebuilding an 8-atom cluster again. This step is summarized in the following Figure:
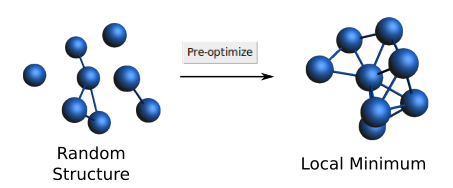
The initial structure is not important, but more iterations may be needed to get the same results. For this tutorial, we suggest starting with the same local minimum to improve reproducibility.
Co -0.57747929 3.09962106 -1.40010667
Co -1.94268805 1.60982864 -0.21835532
Co 0.68130178 -0.28713038 1.61855474
Co 0.41911715 1.21862699 -0.30327160
Co 1.35380790 3.40899190 -0.11445733
Co 1.88623229 1.82216225 1.57360827
Co 0.56876970 -1.15098318 -0.59251345
Co 2.52877908 0.02596128 0.06823723
To setup the Basin Hopping calculation, follow these steps:
 next to the Task to specify the PES Exploration settings.
next to the Task to specify the PES Exploration settings.10.4.
Details on the different settings can be found in the PES exploration page in the AMS driver manual. For the purpose of this tutorial we will leave everything at the default values, except the number of expeditions and the number of explorers per expedition.
We have now finished the setup of the calculation and we are ready to run it:
Co8_BH)We can visualize the explored energy landscape by using AMSmovie:
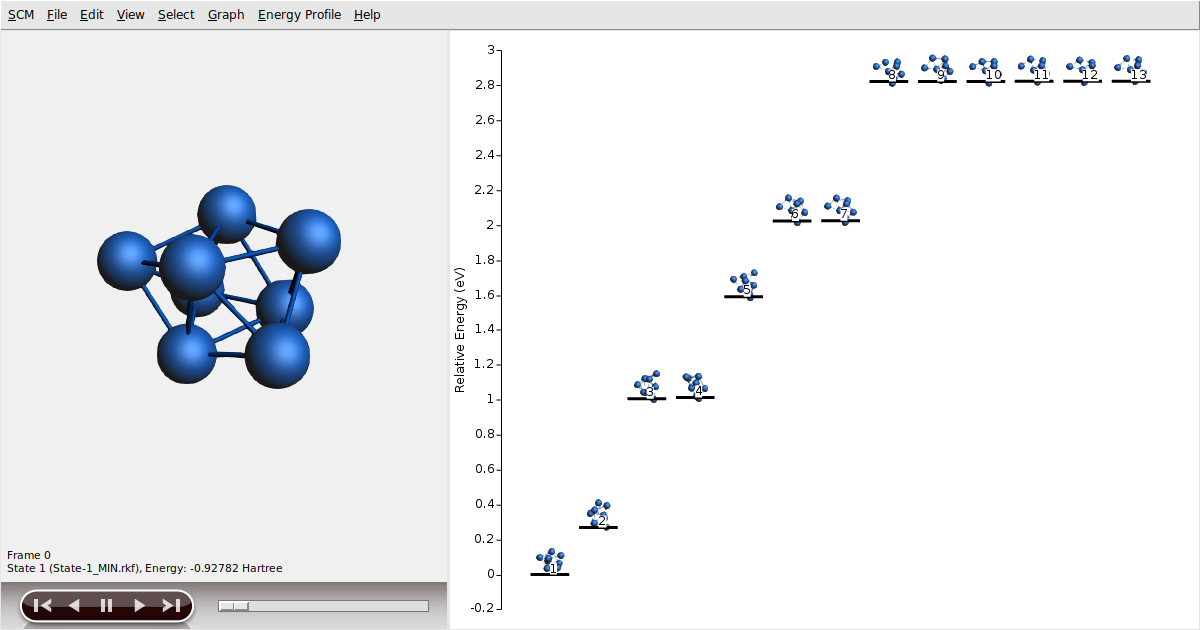
With some luck, the lowest energy state should be at -0.927817 Hartree.
If so, we have caught the global minimum successfully!
Pass the mouse over the state to verify the exact value of the energy.
Basin Hopping optimization is a global optimization that uses random perturbations to jump basins, and a local search algorithm to optimize each basin. So, results may vary given the stochastic nature of the algorithm or differences in numerical precision. Consider running the example a few times and compare the outcomes.
For the next step, we will use the Co8 global minimum. So, copy its geometry and paste it into a new AMSinput.
Growth Mechanism Co8+Co → Co9¶
Firstly, we need the geometry of the Co8 global minimum obtained in the previous section. In case you skipped the previous section, just copy-paste the following coordinates into AMSinput.
Co -0.03403571 1.12611646 -2.08186475
Co -1.53717952 1.78357073 -0.30583453
Co 1.68546231 -0.17156299 1.49980432
Co -0.01541891 1.59507948 1.61007493
Co 0.67964989 2.82625193 -0.46130535
Co 2.34451750 2.13563230 1.20356390
Co -0.20262505 -0.26260499 -0.06462506
Co 1.99747005 0.71459565 -0.76811757
The new AMSinput window does not know yet that we want to perform PES explorations with the ReaxFF engine. Let us quickly restore the engine settings we have used for the Basin Hopping earlier:
→ .Using the drawing tools of AMSinput, add a new Co atom roughly on the cluster’s surface. Then, use the Pre-optimize button to get the closest local minimum, making sure that only the new atom is selected, and only that selection is optimized; we would like to keep the structure of the Co8 intact. Again, the initial structure is not important, but more iterations may be needed to get the same results. We suggest starting with the following coordinates to improve reproducibility. But, we invite you to use the first option to compare times and outcomes.
Co -0.03403571 1.12611646 -2.08186475
Co -1.53717952 1.78357073 -0.30583453
Co 1.68546231 -0.17156299 1.49980432
Co -0.01541891 1.59507948 1.61007493
Co 0.67964989 2.82625193 -0.46130535
Co 2.34451749 2.13563230 1.20356390
Co -0.20262505 -0.26260499 -0.06462506
Co 1.99747005 0.71459565 -0.76811757
Co 3.01465991 2.87039628 -0.97767612
First, we would like to define two regions: 1) adsorbate, corresponding to the added atom, and 2) cluster, corresponding to the Co8 global minimum. Then, we use the defined cluster region to fix the coordinates of the Co8.

In this step, we are interested in exploring all possible processes in which the added atom may be involved. Here, we will use the Process Search subtask for this aim. To clarify, we refer to processes as paths that connect local minima through saddle points (transition states). The subtask Process Search will start in the proposed local minimum and try to find a nearby saddle point starting from a random displacement of the added atom. If a saddle point is found, it will perform a geometry optimization pushing the atom to the other side of the saddle point to find the new local minimum to which the original state is connected. This process is repeated several times, starting from all the local minima discovered so far.
Once the energy landscape is obtained, we will also ask to calculate the associated binding-sites lattice.
Binding sites are defined as the atoms’ position from the adsorbate region which are neighbors of the atoms in the region cluster and associated with a local minimum configuration in the energy landscape.
Additionally, two binding sites are considered connected if the following two requirements are met: 1) there is a transition state connecting two local minima associated to these binding sites (notice that there may be multiple local minima associated with the same binding site), and 2) there is at least an atom (from the adsorbate region) that changes its position from the first binding site to the second one mediated by the same transition state described above.
In this process, AMS will align as much as possible all local minima and transition states to the input file’s structure but ignoring the atoms in the region adsorbate trying to establish a common reference frame.
If the structure of the region cluster changes significantly for different configurations, the binding sites may not be well defined.
For this example, we have frozen the Co8 cluster region, which guarantees that we will not have that issue.
To setup the Process Search calculation, follow these steps:
100.4. next to the Structure Comparison5.0 Åcluster as the Reference region0.5 Å.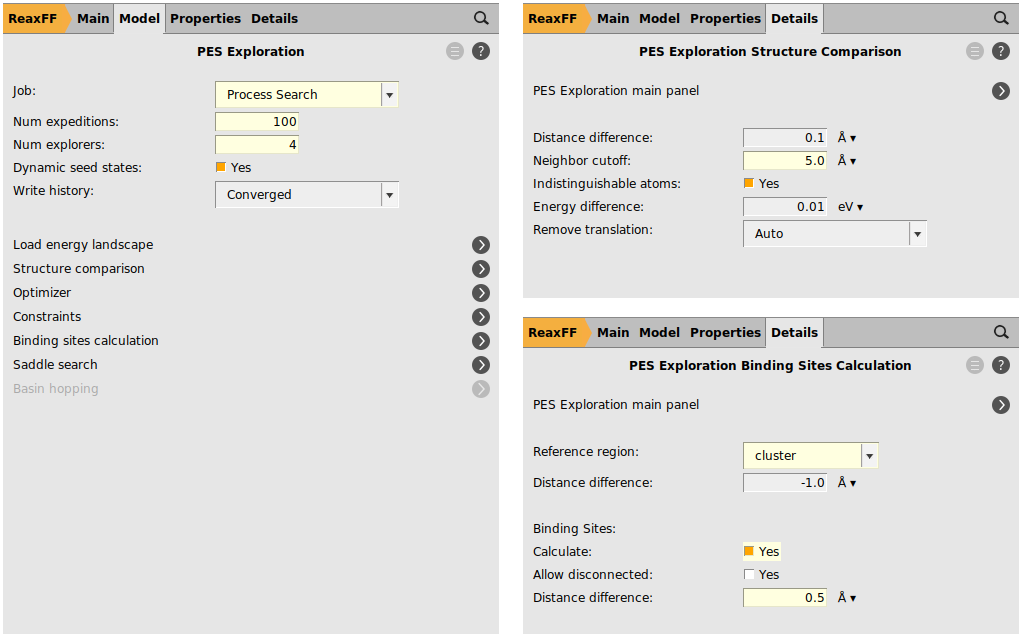
We have now finished the setup of the calculation and we are ready to run it:
Co8+Co_PS)In the AMSinput, we will see the binding-sites lattice superimposed to the cluster’s initial geometry:
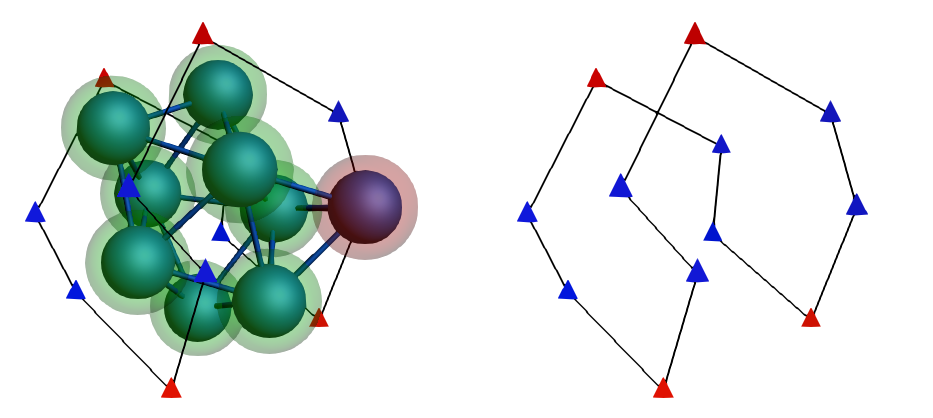
Notice that the added atom is located over one of the binding sites. The symbols used to represent the binding sites are related to the number of atom neighbors (within a 5 Å of distance, see above Neighbors cutoff option) and the colors with the average energy of the contributing states. In our case, because there is only one type of neighborhood, there is only one symbol: triangles.
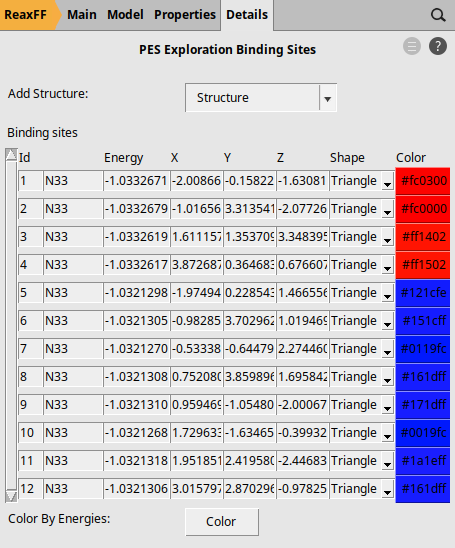
These binding sites have three neighbors in the first coordination shell and three neighbors in the second. Because of this, the labels are N33. Regarding their color, we have two groups: 1) Red for the most stable adsorption-sites with the energy of around -1.0333 Hartree, and 2) Blue for the rest of the binding sites with the energy of -1.0321 Hartree.
These details can be seen from the panel Details → PES Exploration Binding Sites:
We can visualize the explored energy landscape by using AMSmovie:
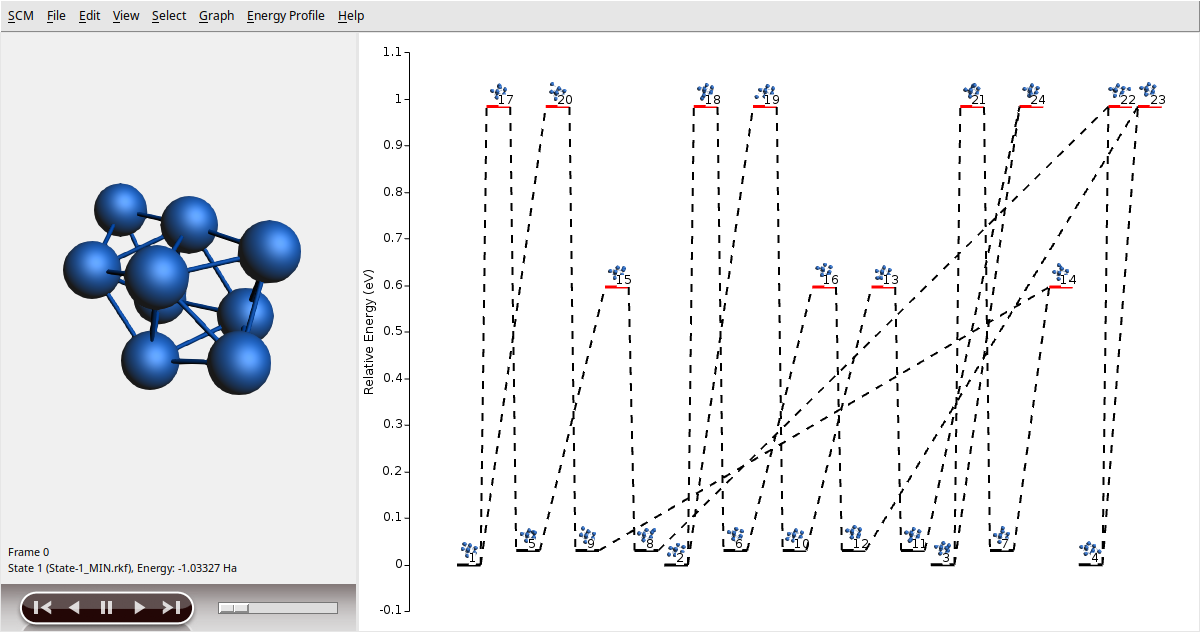
You should get a graphical representation of the states discovered during the searching in the right panel. Minima are shown with a black level, while transition states are shown using a red level. You can move around the different states by clicking on them. The selected state is shown on the left panel to appreciate details.
As a final comment, due to the stochastic nature of the algorithm and/or differences in numerical precision, you can get different results for the energy landscape exploration. In our example, particularly, we have chosen 100 for Num expeditions to guarantee that you can get the full set of critical points; 12 local minima and 12 transition states.
Note
You can see the vibrational spectrum of any state by using right-click on it and then selecting Open in module → Spectra in the popup window. For the present calculation, only the frequencies of the modes related to the unconstrained atoms are available.
Note
You can remove any state by using right-click on it and then selecting Delete level in the popup window.
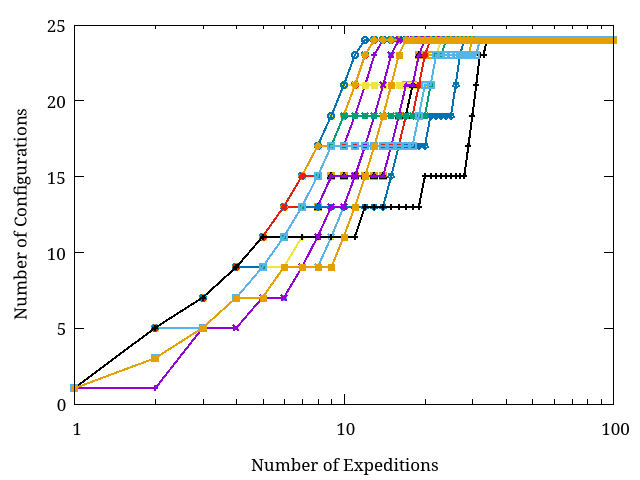
In the Figure below, we show the number of configurations obtained by 20 different runs as the number of expeditions increase. Notice that at least 100 expeditions are required to guarantee you get the 24 configurations.