Multiscale modeling of OLED devices¶
Introduction¶
Starting with the 2022 release of the Amsterdam Modeling Suite we include a set of workflow scripts for multiscale OLED modeling. These workflows are developed and validated in close collaboration with the Eindhoven University of Technology to bridge the gap between ab-initio atomistic modeling of OLED molecules with AMS, and device level kinetic Monte Carlo simulations using our Bumblebee code. We aim to provide a fully integrated multiscale simulation platform for the digital screening and prediction of successful OLED materials and devices.
This tutorial teaches you to:
Deposit a thin film of β-NPB in a simulated physical vapor deposition.
Calculate the distribution of properties such as ionization potential, electron affinity and dipole moments for all molecules in the film.
Transfer the data to the Bumblebee software for OLED device simulations involving your material.

We will use β-NPB as an example compound for this tutorial, an isomer of the common hole transport material α-NPB. Both the deposition as well as the calculation of the properties is computationally quite expensive and requires time as well as a HPC system at your disposal. The full results for both β-NPB and α-NPB can be found in the OLED material database, so you will not have to complete the tutorial in order to be able to look at the results. None of the steps in this tutorial are specific to β-NPB, so you might just follow the tutorial using your own molecule.
See also
For a detailed description of the methods and options, see the OLED workflows page in the manual.
Deposition of the material¶
 →
→ 
 on the right to go to the Model → OLED Deposition panel.
on the right to go to the Model → OLED Deposition panel.
We will have to obtain the 3D structure of the molecule we want to deposit. Our example compound β-NPB can be found through the search box in AMSinput, as can all other molecules from the OLED material database.
 .
.NPB and select beta-NPB from the list.
If you do not have the 3D structure of molecule to deposit yet, you may have to build it using the tools in AMSinput, see the tutorials on building structure. If you can find the SMILES string of the molecule (e.g. from PubChem), you can also just paste that into AMSinput. This will try to generate a 3D structure using RDKit, which should work quite well for organic molecules. No matter how you obtain the initial structure, we suggest you do a quick pre-optimization with GFN1-xTB.
 .
.If you loaded the β-NPB structure through the search box, the pre-optimization will not change much, since all the molecule from the OLED material database have already been optimized with GFN1-xTB. In general this step is important though, as it will also update the bond orders to the ones calculated from the GFN1-xTB orbitals. The deposition is done with the ForceField engine using the UFF4MOF-II force field. It will use the bonds and atom types are they are set up in AMSinput, so it is important to get them right at this point. We should therefore also check the atom-types that will be used:
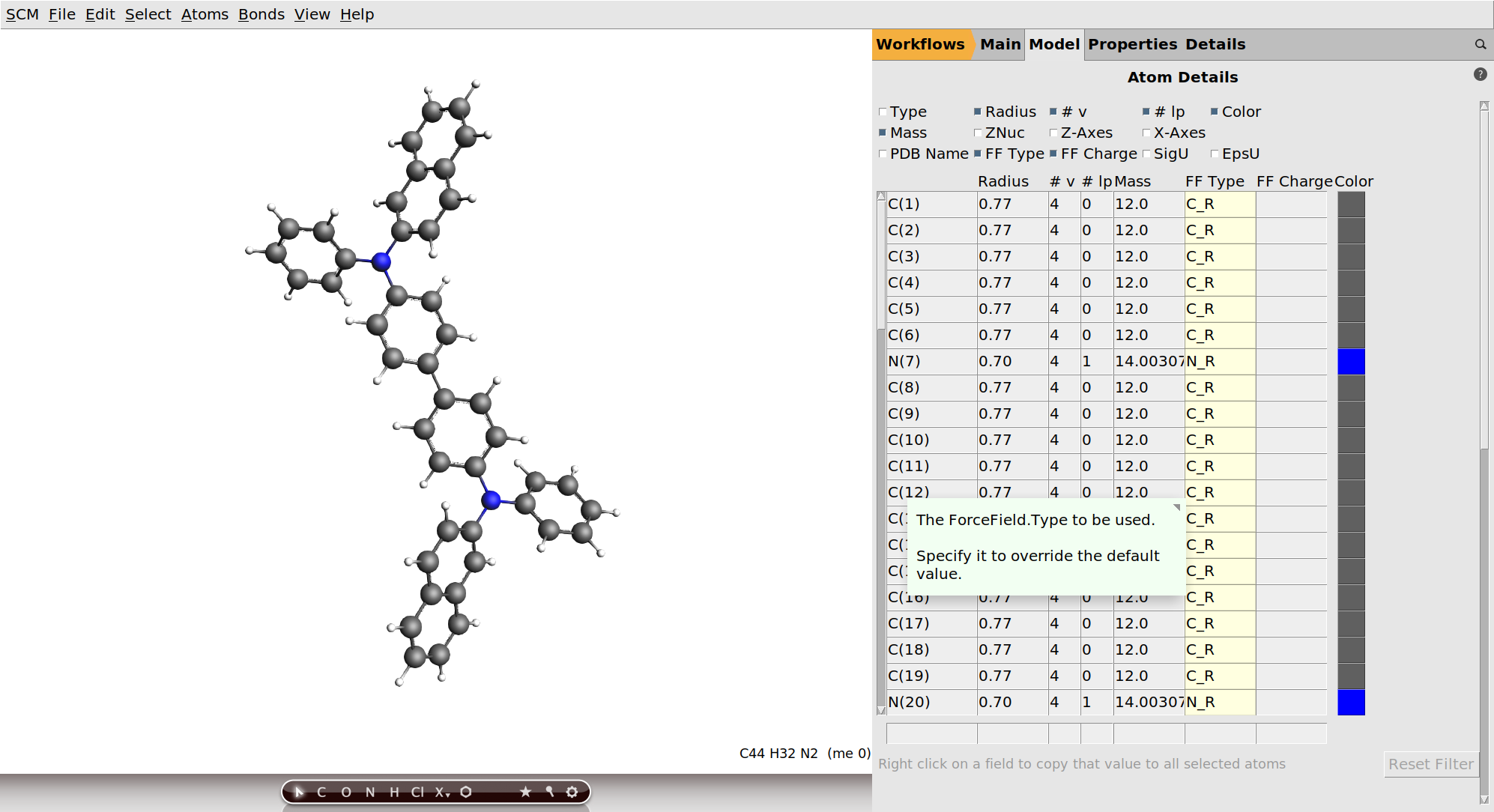
For β-NPB the automatically determined atom types are fine. If we wanted to deposit a mixture, e.g. for the simulation of a host-guest system, we could go back to the Model → OLED deposition panel, click the  button and repeat the process for the other molecules. However, for the purpose of this tutorial we will stick with pure β-NPB.
button and repeat the process for the other molecules. However, for the purpose of this tutorial we will stick with pure β-NPB.
We will not change any of the deposition settings from their default values. By running with all default settings, the results we get should match the β-NPB in the OLED material database.
Tip
You can use a GPU to significantly accelerate the deposition by enabling the LAMMPS offload on the main deposition panel. In order to do so, you will first need to build LAMMPS on the system where you want to run the deposition, see: Setting up LAMMPS.
OMP package.GPU package.
We are now ready to run the calculation.
beta-NPB_deposition.The first step in the deposition workflow is an optimization of the molecule at the DFT level. This is done to determine the atomic charges that will be used for modeling the electrostatics with the force field. Please refer to the OLED workflows manual for a detailed description of the workflows.
You can also monitor the progress of the workflow in AMSmovie. Even though the workflow technically consists of multiple AMS jobs, the GUI is smart enough to open the latest, currently running job when you open AMSmovie.
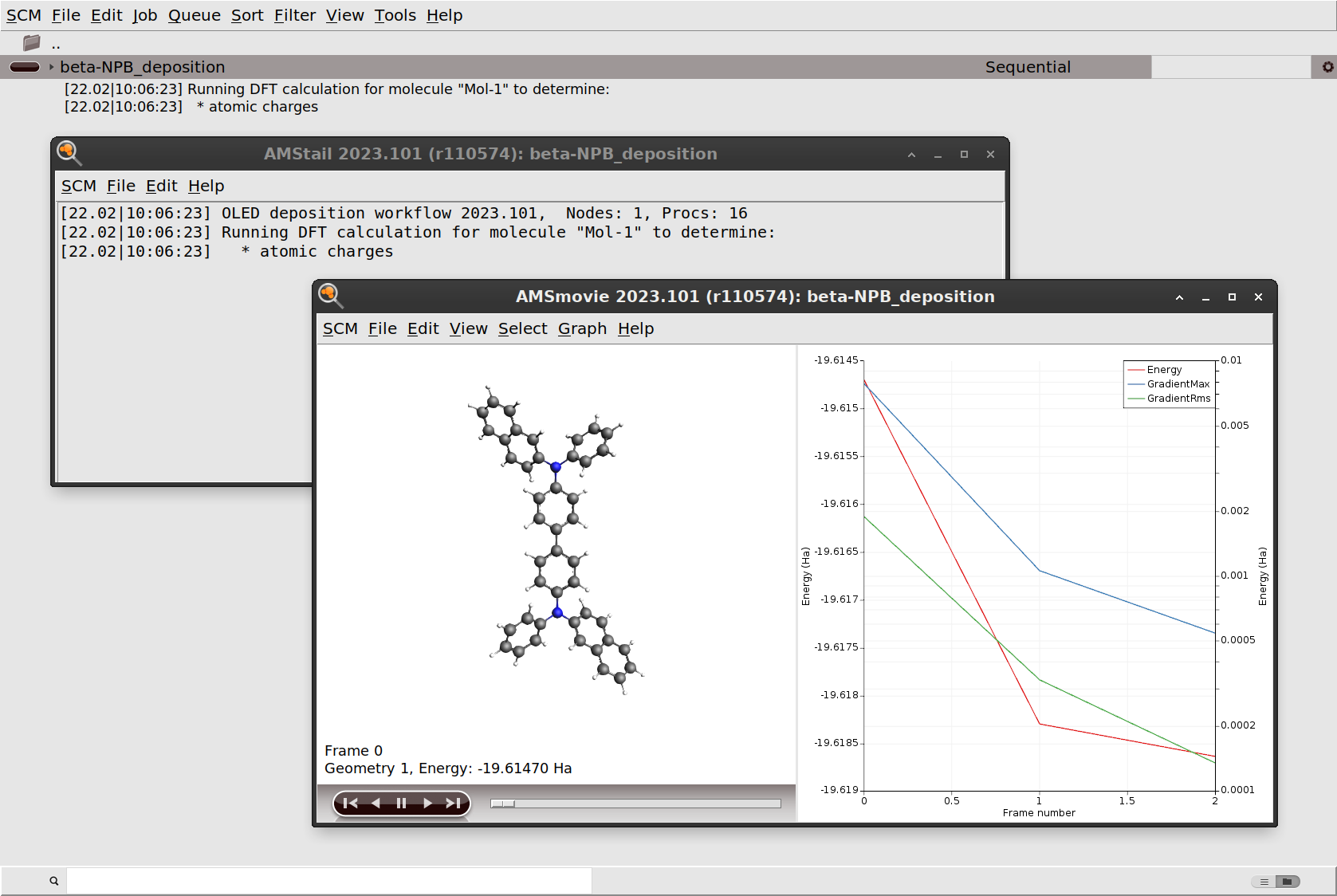
Once the initial DFT calculation has finished, the molecule is equilibrated to the deposition temperature. At this point the workflow script will also print some information about the estimated number of molecules to deposit in order to grow a slab of the desired thickness. After this, the actual simulated physical vapor deposition will begin.
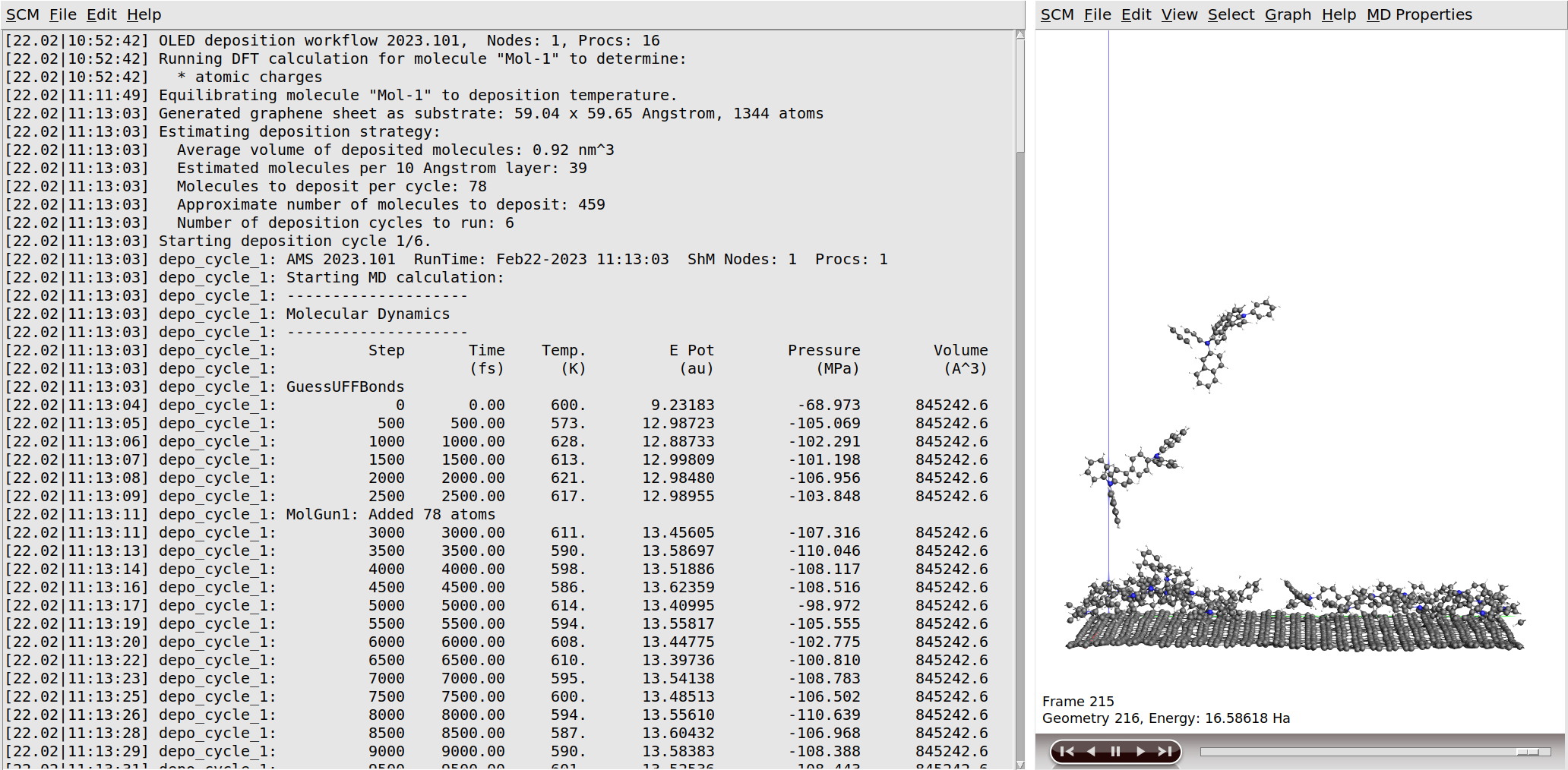
Growing a 120 Å thick layer in the simulated physical vapor deposition will take a few days at least.
We therefore recommend running these jobs using a remote queue. If you have access to a compute cluster, but have not yet set up remote queues for it, we recommend doing this before running real depositions. You can find instructions on how to set up remote queues in the installation guide and the AMSjobs manual.
If you monitor the logfile of the deposition for a while, you will see that the entire deposition is split into a number of cycles. This is done for reasons of computational efficiency, as it allows the periodic trimming of the bottom so that only the growing surface needs to be simulated. Details can be found in the OLED workflows manual. In the end of the simulation the entire system is annealed first annealed down to room temperature and then completely frozen in a geometry optimization. The entire process can be seen in the video on the right.
The output of the deposition workflow is described in detail in the manual. The morphology.in file is the final morphology, which is also stored in the OLED material database as pure/beta-NPB/morphology.in.
You can also download the β-NPB morphology here. The file can be opened in AMSmovie.
Calculation of material properties¶
Once we have obtained the morphology, we can use it to run the OLED properties workflow and obtain material properties such as ionization potential and electron affinity.
→ on the right to go to the Model → OLED Properties panel.The OLED properties workflow has a number of settings, which we will all leave at their default values in this tutorial.
The data from the OLED material database has been generated with the default settings, so we will generate results that can directly be compared to the materials from the database.
The only real input we will need to provide is the morphology itself.
If you did not run through the first part of this tutorial, we suggest you download the β-NPB morphology now or take it from the database, which you can open by clicking the Import button on the right panel.
morphology.in file for β-NPB.
This is all the setup we need. We can now save the job.
beta-NPB_properties.The OLED properties workflow is computationally very expensive, and it is not practical to run it on your local machine: we will need to use some HPC facilities to run these jobs. You can do so from AMSjobs by using a remote queue. You can find instructions on how to set up remote queues in the installation guide and the AMSjobs manual. The OLED properties workflow consists of many independent chains of calculations for the individual molecules in the morphology, making it very scalable up to hundreds of CPU cores.
Note
Multi-node OLED properties jobs are currently only supported for SLURM based clusters. For other batch systems, you will be limited to running the jobs on a single node.
Once you have configure a suitable remote queue for your job, we are ready to run.
beta-NPB_properties job.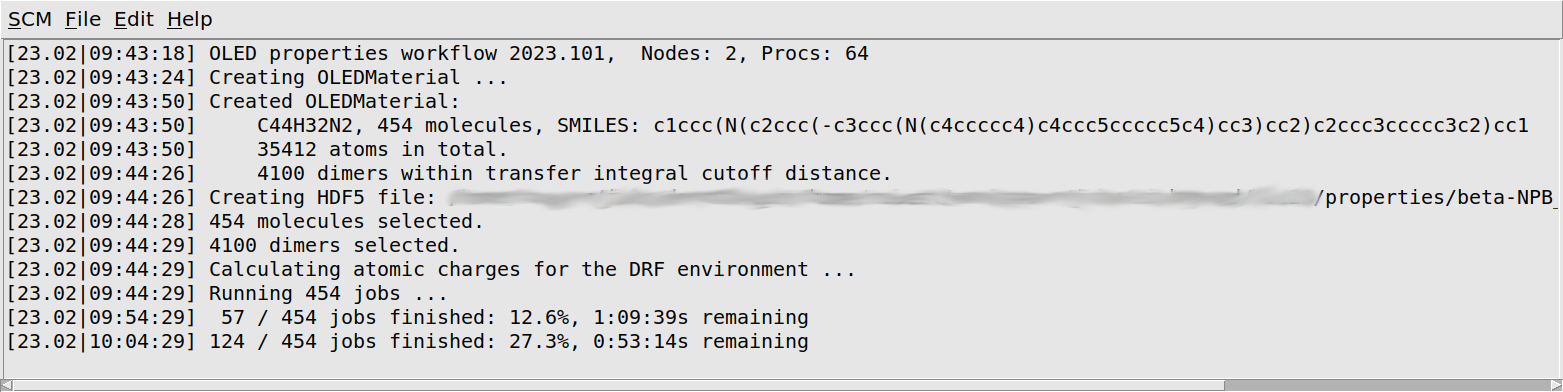
You will first see some information about your morphology printed: the number of molecules of each separate species (there is just one species for our pure β-NPB morphology), as well as the total number of atoms, and the number of dimers considered in the calculation of the transfer integrals.
The workflow script will periodically print a progress update. You will see that it loops over all molecules twice: first to quickly calculate the atomic charges that will later be used as the environment, and then again to calculate the properties of each molecule in its environment. Finally there is a loop over dimers of neighboring molecules, which calculates the transfer integrals. Depending on the machine you are running on, it may take a few days to run the calculations for all molecules.

When the job has finished, it will print a quick summary of the most important results.
The workflow script will also produce a small HDF5 file.
The HDF5 file contains a summary of the results. It can be opened in AMSview or analyzed with our Python environment.
(If you do not want to wait for the calculation to finish, you can download the file here or take it from the OLED material database, where it is stored as pure/beta-NPB/properties.hdf5.)
You will be greeted by a view of a box that is filled with disconnected spheres. Note that these spheres do not represent atoms, but individual molecules from the morphology. If we had deposited a host-guest system, the spheres representing host and guest molecules would have a different color. We can now visualize the properties we calculated via the Properties menu in the top bar.
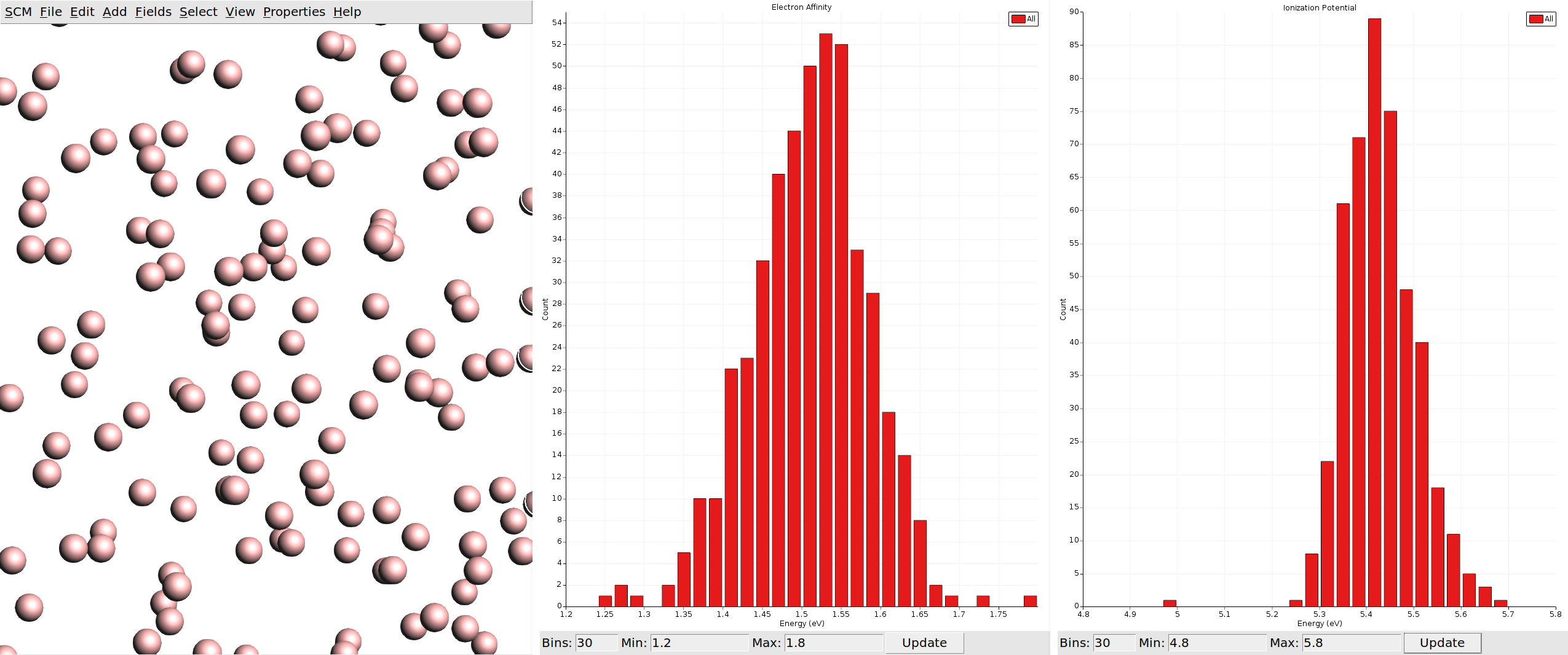
We can also visualize vector properties as arrows overlaid onto the spheres, and plot histograms of the vector magnitudes as well as angles against the z-axis (the growth direction of the thin film in the deposition).

Detailed analysis of the data from the HDF5 file is best done from Python. See the manual section for details.
The HDF5 file can be used as input for Bumblebee simulations of OLED devices. Please refer to the Bumblebee documentation for details on the construction of virtual OLED stacks using the materials obtained by the OLED workflows.