DFTB
Fast approximate DFT for molecules, 1D, 2D and 3D
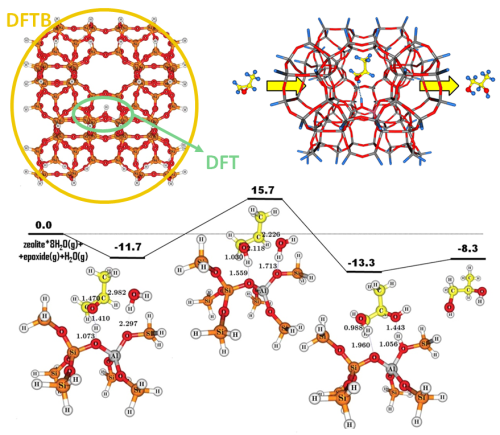
Density-Functional based Tight-Binding (DFTB) allows to perform calculations of large systems over long timescales even on a desktop computer. Relatively accurate results are obtained at a fraction of the cost of density functional theory (DFT) by using pre-calculated parameters, a minimal basis, and including only nearest-neighbor interactions. Long-range interactions are described with empirical dispersion corrections, while third-order corrections accurately handle charged systems.
The DFTB module can treat molecular as well as periodic systems (1D for nanotubes, 2D for surfaces, 3D for bulk), and as such can be used as a fast pre-optimizer for full molecular and periodic DFT calculations with ADF and BAND. The DFTB license also includes the semi-empirical MOPAC library, which uses similar approximations and is parametrized against experimental heats of formations.
With the integrated graphical user interface it is easy to run, set up and analyze DFTB jobs, or to use DFTB as a quick pre-optimization tool.
Visualizing DFTB molecular orbitals and interaction diagrams
Ole Carstensen shows how you can easily visualize MOs with DFTB, and get insight in the orbital interactions by defining fragments for the MO levels diagram.