Intrinsic Reaction Coordinate (IRC)¶
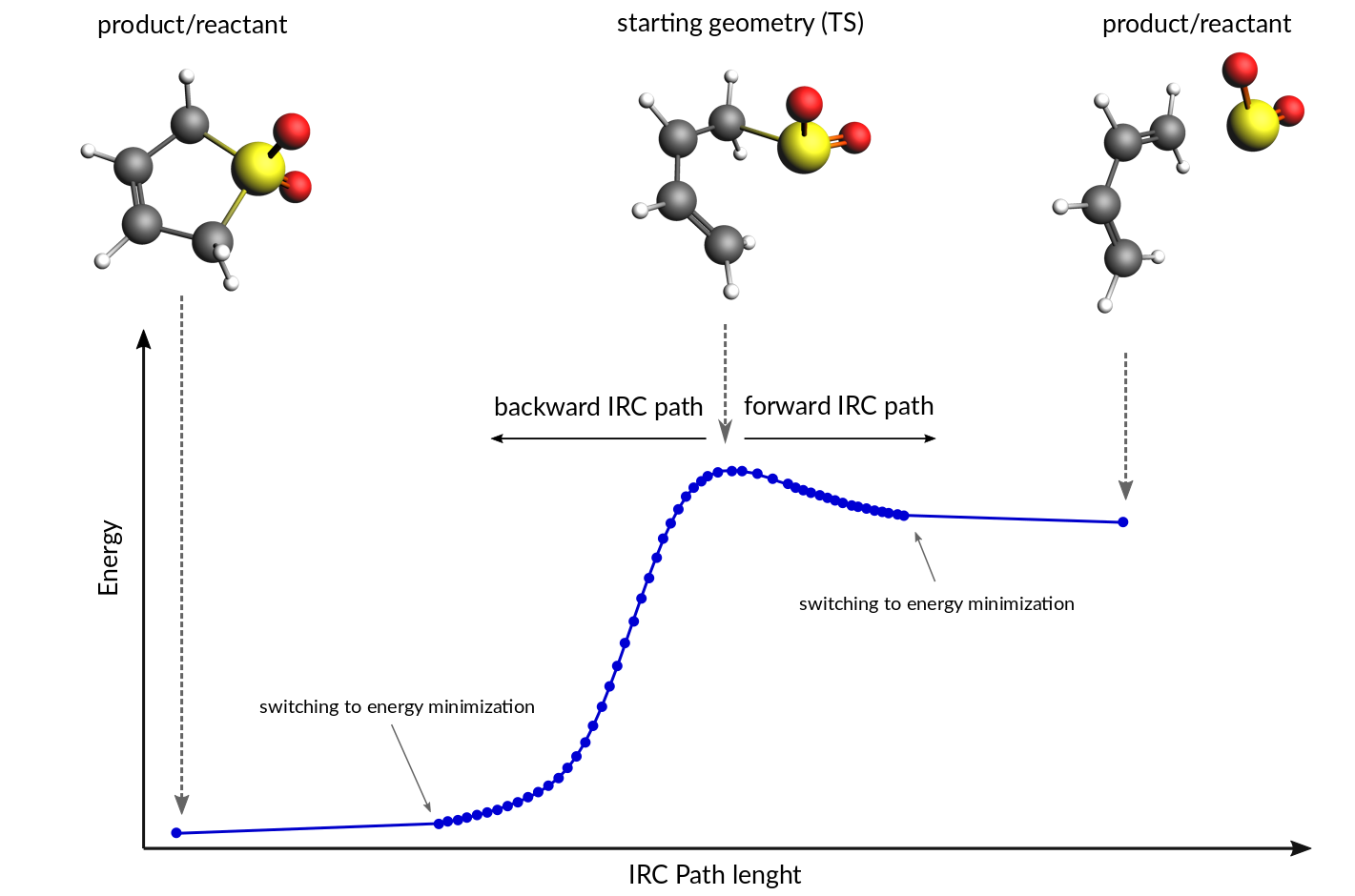
The path of a chemical reaction can be traced from the transition state (TS) to the products and/or reactants using the Intrinsic Reaction Coordinate (IRC) method [1] [2]. The method assumes that the starting geometry is a fair approximation of the TS. A minimum energy profile (MEP) is defined as the steepest-descent path on the potential energy surface from the transition state down towards a local minimum. An IRC path is defined similarly but in the mass-weighted coordinates [3], which means that instead of the steepest descent direction it follows that of the maximum instantaneous acceleration. This makes IRC somewhat related to the Molecular Dynamics method. The energy profile is obtained as well as the length and curvature properties of the path, providing the basic quantities for an analysis of the reaction path.
See also
Examples and the PES exploration for hydrohalogenation tutorial
Method details¶
Calculation of an IRC path consists of two nested loops, the so-called outer and inner loops. The outer loop runs over IRC points and the inner loop is over geometry optimization steps for the given IRC point. The first IRC point starts from the transition state geometry, which is a saddle point, in one of the two possible downhill directions. Each IRC point after that starts from the optimized geometry of the previous point. At the start of every step, the pivot point is determined, which is a point at the Step/2 distance in the direction opposite to the gradient. When working in the mass-weighted coordinates, this direction corresponds to the acceleration of the corresponding atom. The final point of the given IRC step corresponds to the energy minimum point at the same distance (Step/2) from the pivot point further downhill. More precisely, the coordinates of the target point are optimized during the inner loop to minimize projection of the gradient on the hypersphere of radius Step/2 around the pivot point. The angle between the (pivot-start) and (pivot-final) vectors determines the curvature of the reaction path. If this angle becomes smaller than 90 degrees then the IRC scan is considered to have reached vicinity of an endpoint and the program switches to energy minimization (options for this energy minimization can be specified in the Geometry Optimization block.). If the angle is between 90 and 120 degrees then the current IRC step is canceled and a new one is started from the same starting point with half the initial Step parameter. In all other cases the optimized geometry becomes a starting point for the next IRC step. By default, when the forward path is completed the backward one is started from the same TS geometry. When both forward and backward paths are complete, a summary of the whole reaction path is printed to the output.
Input¶
The IRC scan in AMS is triggered by setting the Task to IRC:
Task IRC
All IRC-related options are specified in the IRC input block:
IRC
Convergence
Gradients float
Step float
End
CoordinateType [Cartesian | Delocalized]
Direction [Both | Forward | Backward]
InitialHessian
File string
Type [Calculate | FromFile]
End
KeepConvergedResults Yes/No
MaxIRCSteps integer
MaxIterations integer
MaxPoints integer
MinEnergyProfile Yes/No
MinPathLength float
Restart
File string
RedoBackward integer
RedoForward integer
End
Step float
End
All keys of the IRC block have reasonable defaults or are optional. Thus, in principle, the IRC block can be omitted altogether. These are some of the main options:
IRCDirectionType: Multiple Choice Default value: Both Options: [Both, Forward, Backward] Description: Select direction of the IRC path. The difference between the Forward and the Backward directions is determined by the sign of the largest component of the vibrational normal mode corresponding to the reaction coordinate at the transition state geometry. The Forward path correspond to the positive sign of the component. If Both is selected then first the Forward path is computed followed by the Backward one. StepType: Float Default value: 0.2 GUI name: Step size Description: IRC step size in mass-weighted coordinates, sqrt(amu)*bohr. One may have to increase this value when heavy atoms are involved in the reaction, or decrease it if the reactant or products are very close to the transition state. InitialHessianType: Block Description: Options for initial Hessian at the transition state. The first eigenvalue of the initial Hessian defines direction of the first forward or backward step. This block is ignored when restarting from a previous IRC calculation because the initial Hessian found in the restart file is used. FileType: String GUI name: File Description: If ‘Type’ is set to ‘FromFile’ then in this key you should specify the RKF file containing the initial Hessian (or the ams results dir. containing it). This can be used to load a Hessian calculated previously with the ‘Properties%Hessian’ keyword. If you want to also use this file for the initial geometry then also specify it in a ‘LoadSystem’ block. TypeType: Multiple Choice Default value: Calculate Options: [Calculate, FromFile] GUI name: Initial Hessian Description: Calculate the exact Hessian for the input geometry or load it from the results of a previous calculation.
The following keys set limits on the number of steps for the inner and outer IRC loops and, related to that, the geometry optimization criteria. Note that tighter criteria may require a greater MaxIterations limit. Please also note that the outer loop limits are valid for each half of the path (forward and backward) separately. That is, if all settings are left at their defaults then up to 200 IRC points may be calculated, each of them may require up to 300 energy evaluations.
IRCMaxIRCStepsType: Integer GUI name: Maximum IRC steps Description: Soft limit on the number of IRC points to compute in each direction. After the specified number of IRC steps the program will switch to energy minimization and complete the path. This option should be used when you are interested only in the reaction path area near the transition state. Note that even if the soft limit has been hit and the calculation has completed, the IRC can still be restarted with a ‘RedoBackward’ or ‘RedoForward’ option. MaxPointsType: Integer Default value: 100 GUI name: Maximum points Description: Hard limit on the number of IRC points to compute in each direction. After the specified number of IRC steps the program will stop with the current direction and switch to the next one. If both ‘MaxPoints’ and ‘MaxIRCSteps’ are set to the same value then ‘MaxPoints’ takes precedence, therefore this option should be used to set a limit on the number of IRC steps if you intend to use the results later for a restart. MaxIterationsType: Integer Default value: 300 GUI name: Maximum iterations Description: The maximum number of geometry iterations allowed to converge the inner IRC loop. If optimization does not converge within the specified number of steps, the calculation is aborted. ConvergenceType: Block Description: Convergence at each given point is monitored for two items: the Cartesian gradient and the calculated step size. Convergence criteria can be specified separately for each of these items. The same criteria are used both in the inner IRC loop and when performing energy minimization at the path ends. GradientsType: Float Default value: 0.001 Unit: Hartree/Angstrom GUI name: Gradient convergence Description: Convergence criterion for the max component of the residual energy gradient. StepType: Float Default value: 0.001 Unit: Angstrom GUI name: Step convergence Description: Convergence criterion for the max component of the step in the optimization coordinates.
MinPathLengthType: Float Default value: 0.1 Unit: Angstrom Description: Minimum length of the path required before switching to energy minimization. Use this to overcome a small kink or a shoulder on the path.
The following keys modify other aspects of the IRC scan:
IRCCoordinateTypeType: Multiple Choice Default value: Cartesian Options: [Cartesian, Delocalized] GUI name: Coordinates used for optimization Description: Select the type of coordinates in which to perform the optimization. Note that the Delocalized option should be considered experimental. MinEnergyProfileType: Bool Default value: No GUI name: Minimum energy profile Description: Calculate minimum energy profile (i.e. no mass-weighting) instead of the IRC. KeepConvergedResultsType: Bool Default value: Yes Description: Keep the binary RKF result file for every converged IRC point. These files may contain more information than the main ams.rkf result file.
It is possible to restart an IRC calculation that crashed, has been killed or exceeded the MaxPoints limit, or to re-compute the path starting from a certain point, using the Restart key:
IRCRestartType: Block Description: Restart options. Upon restart, the information about the IRC input parameters and the initial system (atomic coordinates, lattice, charge, etc.) is read from the restart file. The IRC input parameters can be modified from input. Except for ‘MaxPoints’ and ‘Direction’ all parameters not specified in the input will use their values from the restart file. The ‘MaxPoints’ and ‘Direction’ will be reset to their respective default values if not specified in the input. By default, the IRC calculation will continue from the point where it left off. However, the ‘RedoForward’ and/or ‘RedoBackward’ option can be used to enforce recalculation of a part of the reaction path, for example, using a different ‘Step’ value. FileType: String GUI name: Restart Description: Name of an RKF restart file generated by a previous IRC calculation. Do not use this key to provide an RKF file generated by a TransitionStateSearch or a SinglePoint calculation, use the ‘LoadSystem’ block instead. RedoBackwardType: Integer Default value: 0 Description: IRC step number to start recalculating the backward path from. By default, if the backward path has not been completed then start after the last completed step. If the backward path has been completed and the ‘RedoBackward’ is omitted then no point on the backward path will be recomputed. RedoForwardType: Integer Default value: 0 Description: IRC step number to start recalculating the forward path from. By default, if the forward path has not been completed then start after the last completed step. If the forward path has been completed and the ‘RedoForward’ is omitted then no point on the forward path will be recomputed.
Output¶
A summary of reaction path is printed to the output file at the end of the IRC calculation.
The IRC reaction path can be visualized using the AMSmovie GUI module.
Results of an IRC calculation are also stored in the History section of the ‘ams.rkf’ file, just like in a normal geometry optimization. In addition to the standard KF variables such as “Coords” and “Energy”, the following IRC-specific variables are also created:
- IRCDirection - IRC direction to which this point belongs: 1 - forward, 2 - backward.
- IRCIteration - the IRC (a.k.a. the outer loop) iteration number.
- OptIteration - the geometry optimization (a.k.a. the inner loop) iteration number (0 means the results correspond to the converged geometry at this IRC step).
- IRCGradMax - value of the max component of the IRC gradient that determines convergence of the inner loop.
- IRCGradRms - the RMS value of the IRC gradient that determines convergence of the inner loop. Both the ircGradRms and the ircGradMax are given in the mass-weighted atomic units for IRC steps and in the atomic units for the final minimization loop.
- ArcLength - length, in Angstrom, of the arc that connects the initial and the final point of this IRC step. The corresponding pivot point is located near the the middle point of the arc.
- Angle - value of the angle (in degrees) between lines connecting the pivot point with the initial and final points. A value of 180 degrees means the path is passing straight through the pivot point, while a smaller value means the path makes a bend at this point.
- PathLength - sum of the ArcLength values from the transition state up to this point, in Angstrom.
- Converged - a Fortran logical value containing the convergence status of the given geometry.
The IRC section of the RKF file contains all the data needed for a successful restart procedure.
References
| [1] | L. Deng, T. Ziegler and L. Fan, A combined density functional and intrinsic reaction coordinate study on the ground state energy surface of H2 CO, Journal of Chemical Physics 99, 3823 (1993) |
| [2] | L. Deng and T. Ziegler, The determination of Intrinsic Reaction Coordinates by density functional theory, International Journal of Quantum Chemistry 52, 731 (1994) |
| [3] | C. Gonzalez and H.B. Schlegel, Reaction Path Following In Mass-Weighted Internal Coordinates J. Phys. Chem. 94, 5523-5527 (1990) |