Work functions at interfaces¶
This tutorial will show how to use BAND to calculate the work function Φ of the
- Al(100)/vacuum interface
- Al(100)/LiF(100) interface

The adsorption of LiF(100) will decrease the work function, as compared to vacuum. The results will be compared to plane-wave-DFT results by Prada S., et al. [1]
| Prada et al. (eV) | This tutorial (eV) | |
| Al(100) Φ | 4.37 | 4.44 |
| Al(100)/LiF(100) ΔΦ | -0.47 | -0.42 |
Al(100)/vacuum¶
 →
→ 
Now generate the Al(100) slab:
Download the Al100.xyz file
Select File → Import Coordinates and select the downloaded file
See also
If you are not yet familiar with the editing tools in AMSinput, take a look at our Introduction to Building structures.
2
Then, set the BAND settings:

 next to K-space
next to K-space7 7
0.002 hartree
This sets up a 7x7 k-space grid. The integration, spline zlm fit, and electronic temperature options help with the SCF convergence.
Save and run the job.
When the job has finished, the Fermi energy is printed at the bottom of the logfile (SCM → Logfile):
<Jul11-2022> <11:41:08> FERMI ENERGY: -0.1632 A.U.
<Jul11-2022> <11:41:08> -4.4412 E.V
Thus, the work function Φ = 4.44 eV. This compares well to the value of 4.37 eV reported by Prada et al.
Note
The Fermi energy only matches the work function for 0D, 1D, and 2D-periodic metal systems (not 3D-periodic bulk systems).
If you use Quantum ESPRESSO (which only supports slab calculations under 3D-periodic boundary conditions, i.e., with a vacuum gap), you need to also calculate the vacuum level by plane-averaging the electrostatic potential. The work function is then the difference between the Fermi energy and the vacuum level. See quantum-espresso.org for more details.
Tip
Get the Fermi energy using the following script that you can run with $AMSBIN/amspython:
from scm.plams import *
results_dir = '/path/to/jobname.results'
job = AMSJob.load_external(results_dir)
fermienergy = job.results.readrkf('DOS', 'Fermi Energy', file='engine')
fermienergy *= Units.convert(1.0, 'hartree', 'eV')
print(f"Job: {results_dir}")
print(f"Fermi energy: {fermienergy:.2f} eV")
Al(100)/LiF(100)¶
When setting up a solid-solid interface, the lattice constants must match. Both Al and LiF are cubic, so if their lattice constants match, also the (100) surface lattice constants will match. At least one of the materials needs to be a bit strained. Here, we will strain the LiF slab to match the Al slab. We will place the LiF slab at a distance of 3.27 Å from the Al slab.
Note
For other interfaces, you may need to apply surface rotations and use surface supercells if the lattice constants of the two materials are very different, in order to not apply too much strain to one of the materials.
Download the LiF-on-Al.xyz file
Select File → Import Coordinates and select the downloaded file
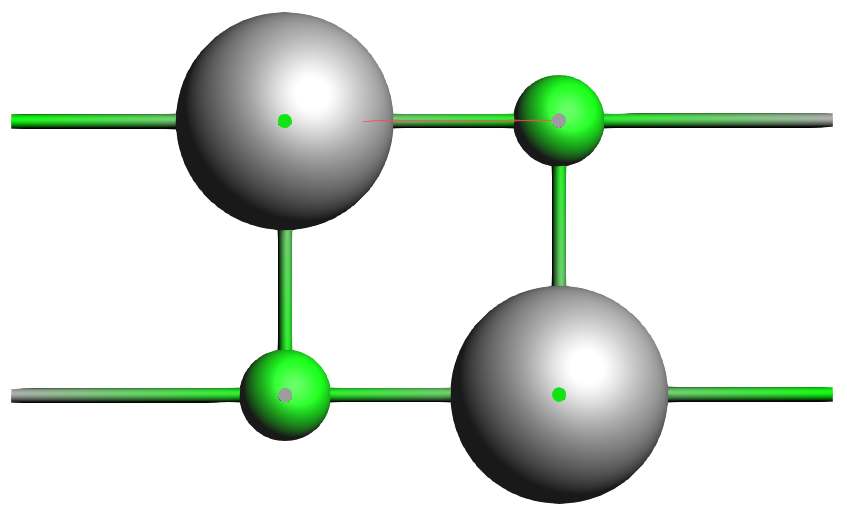
We now want to position this LiF slab on top of the Al slab such that the F atom is 3.27 Å directly above the top Al atom

3.27 Å to first atom (the first atom is the first selected F atom)
This has created the desired interface geometry.

Important
Use the same calculation settings as for the clean Al slab, especially the same number of k-points and functional.
The bottom of the logfile reads:
<Jul11-2022> <11:57:03> FERMI ENERGY: -0.1476 A.U.
<Jul11-2022> <11:57:03> -4.0163 E.V
So the work function Φ = 4.02 eV, giving ΔΦ = (4.02-4.44) = -0.42 eV, which compares well to the value given by Prada et al.: ΔΦ = -0.47 eV (for a 3-layer LiF(100) slab on Al(100)).
| [1] |
|