NiO and DFT+U¶
This tutorial will show you how to perform a single point calculation with the DFT+U formalism using the BAND.


Step 2: Setup the system - NiO¶
You can copy-paste the following information into the AMSinput directly.
2
Ni 0.000 0.000 0.000
O 2.085 2.085 2.085
VEC1 0.000 2.085 2.085
VEC2 2.085 0.000 2.085
VEC3 2.085 2.085 0.000
By default, only the central unit cell is shown. To see a few unit-cell repetitions:
- Click on View → Periodic → Repeat Unit Cells
Step 3: BP86 without Hubbard¶
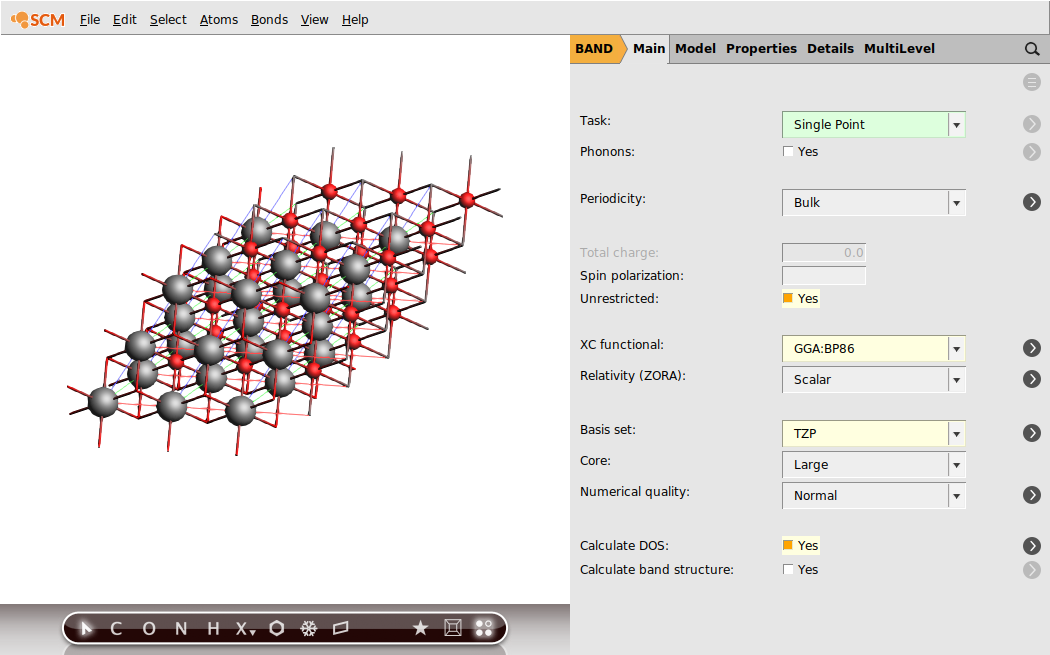
Change the calculation setup (Unrestricted, XC functional, basis set) as follows:
- 1. Check the Unrestricted box.2. Set XC functional to GGA:BP86.3. Set Basis Set to TZP4. Tick the checkbox DOS
Step 3a: Run the calculation¶
Now you can save and run the calculation.
- File → Save, give it a name and press Save.File → Run
Step 3b: Checking the results¶
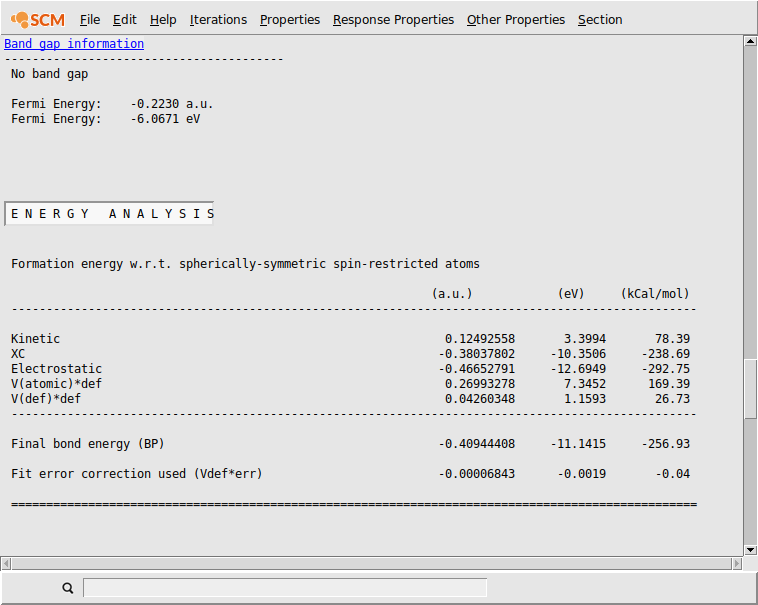
After the calculation finished, you can check the Output for the ‘Band Gap Info’.
- SCM → OutputProperties → Band Gap Info
One can see that there is no band gap at all. This contradicts experimental studies, which predict values between 3.7 to 4.3 eV.
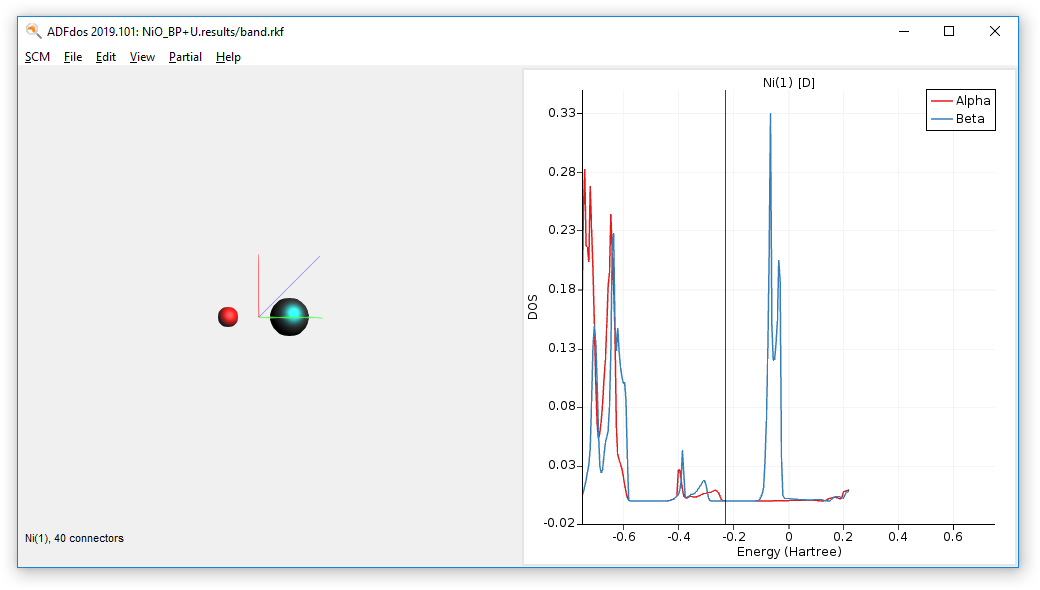
Plotting and examining the partial density of states (pDOS) for this calculation reveals that the d-orbital contributions of Ni are crossing the Fermi level.
- SCM → DOSSelect the Ni atom.Choose Partial → Ni(1) → D-DOS
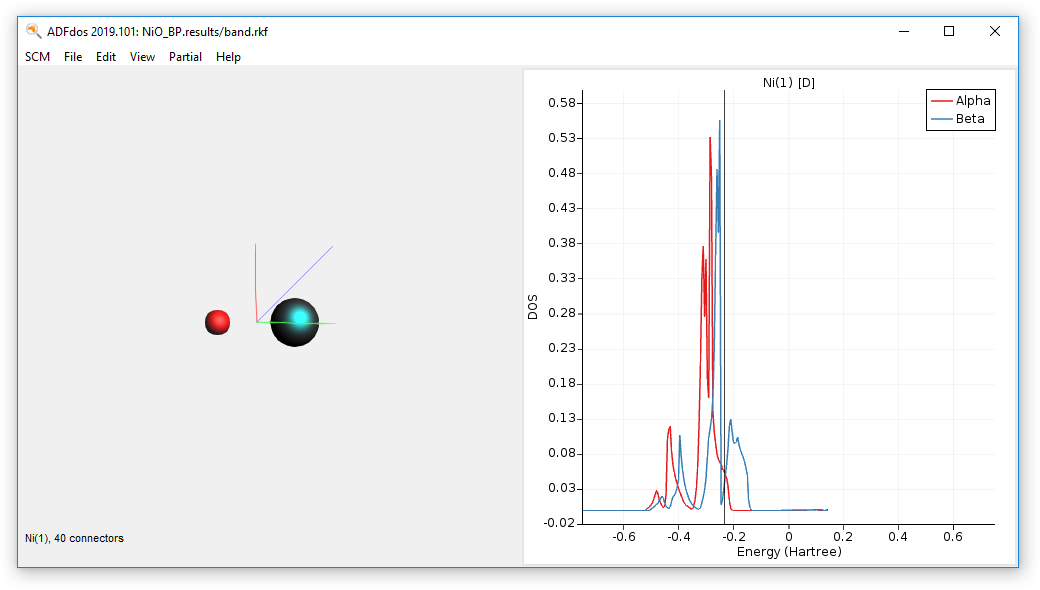
Step 4: Run the calculation - BP86+U¶
Go back to the Main menu of amsinput, change to HubbardU menu, and apply an U value of 0.6 a.u. to the d-orbitals of the Ni atom.
- Go to Model → HubbardU.Set for Ni the l-value to d and the U value to 0.6.
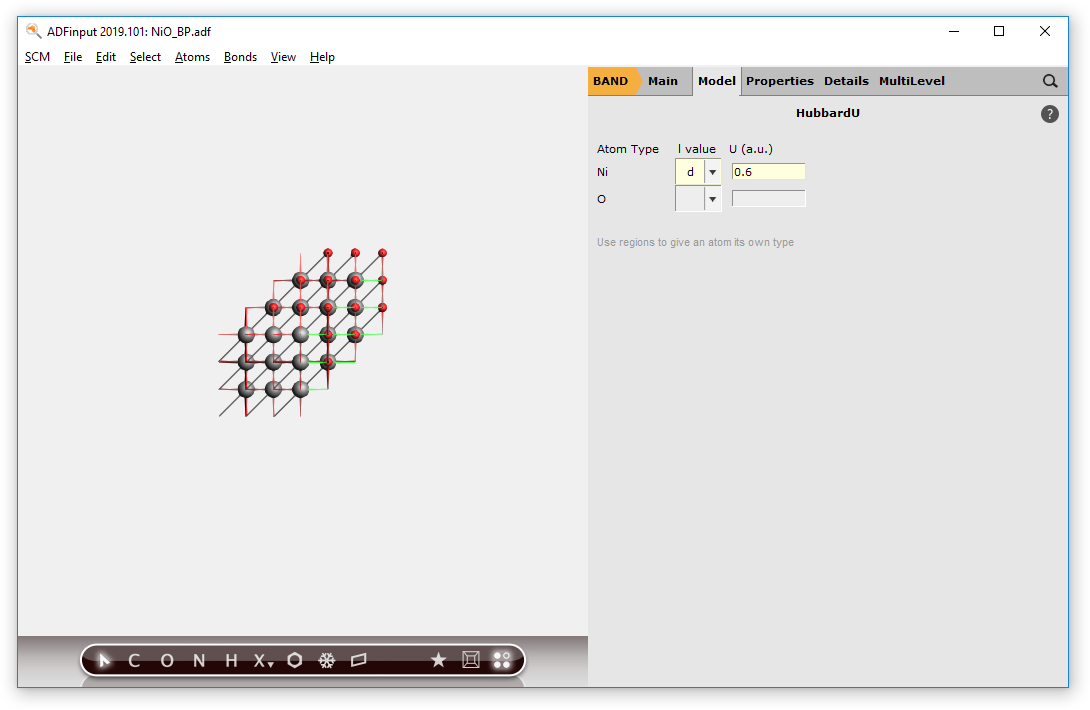
This will influence the Hamiltonian and results in a state which tries to omit partial occupation or degeneracy w.r.t. the d-orbitals.
Step 4a: Run the calculation¶
Now you can save and run the calculation.
- File → Save, give it a name and press Save.File → Run
Step 4b: Checking the results¶
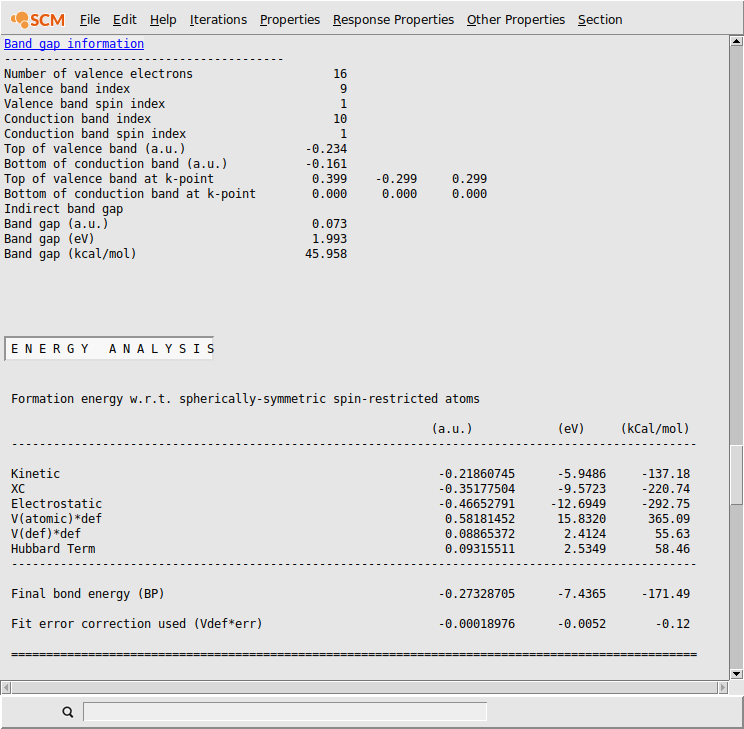
After the calculation finished, you can check the Output for the ‘Band Gap Info’.
- SCM → OutputProperties → Band Gap Info
One can see that there is now a band gap of around 2 eV. This is still less than the experimental values. That can be traced back to the neglection of the correct magnetic behavior of NiO.
Plotting and examining the partial density of states (pDOS) for this calculation reveals that the d-orbital contributions of Ni are no longer crossing the Fermi level.
- SCM → DOSSelect the Ni atom.Choose Partial → Ni(1) → D-DOS