2.3. Import training data (GUI)¶
This tutorial will show you all the ways that you can import finished reference calculations (or results from experiment) into the ParAMS GUI.
For how to do the same using scripting, see Import training data (Python).
There are several ways to import reference data:
- Choosing Job → Add to ParAMS (Ctrl-T) in AMSjobs or Jobs → Add Job (choosing an ams.rkf, OUTCAR, or quantum-espresso.out file) in the ParAMS GUI. This brings up a ResultsImporter dialog and is the recommended way to import a job into ParAMS. In the dialog you can directly import results like energies, forces, stress tensor, charges, bond scans, angle scans, volume scans, and any other PES scan. It is recommended to use the dialog to import those results.
- Choosing File → Add to ParAMS from AMSinput or AMSmovie. This will import the structure (and if using AMSinput also some calculation settings).
- Selecting a previously imported job in the ParAMS GUI and choosing Training Set → Add → Bonds (or similar). This is the recommended way to import geometric data like bond lengths, angles, dihedrals, cell lengths, and cell volume. You can also use it for importing experimental reference results.
Important
Importing geometric data only makes sense for jobs with Task (for new job) Geometry Optimization.
- Choose preferred units
- Reference calculation #1: Geometry optimization of a water molecule
- Charges and forces
- Bonds and angles
- PES scans: bond scan, angle scan, or volume scan
- Energies
- Import a molecular dynamics trajectory
- Import a structure and settings from AMSinput
- Import VASP or Quantum ESPRESSO calculations
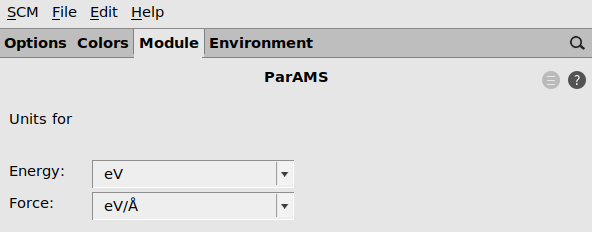
2.3.1. Choose preferred units¶
ParAMS supports several commonly used units, so that you can work with the units that are common in your field.
To set preferred units:
- SCM → PreferencesModule → ParAMSSelect your preferred units for Energy and Forces (for example eV and eV/Å).
2.3.2. Reference calculation #1: Geometry optimization of a water molecule¶
- Open AMSinput and switch to the
 panelSet Task → Geometry OptimizationDraw a water molecule. It is good if it is a bit distorted.Save with the name
panelSet Task → Geometry OptimizationDraw a water molecule. It is good if it is a bit distorted.Save with the namewater.amsRun the job


2.3.3. Charges and forces¶

- This brings up a dialog with a few different options.Set Use ResultsImporter to Add Single Job. This will add a single job to the job collection, based on the final structure of the geometry optimization.Set Task (for new job) to SinglePoint.Set Properties to charges and forces. Note: You can select several entries in the drop-down list!Click Import

In the ParAMS GUI, you can now see

- On the Jobs panel is a job called
waterwith TaskSinglePoint. - On the Training Set panel are two entries
ChargesandForcesfor the jobwater. - On the Engines panel is a new entry
forcefield;<emptySettings>;. This contains the settings used for the reference calculation.
Tip
Select a job in the Jobs panel or a training set entry in the Training Set panel, and switch to the Info panel on the bottom to see some details. To edit the details, double-click in the Details column in the upper table.
Tip
On the Training Set panel, the W column contains the weight of the training set entry. You can edit it in the table. The Value column contains shows the minimum and maximum value.
2.3.4. Bonds and angles¶
To train to geometry data like bonds or angles, you should set the Task (for new job) to Geometry Optimization when importing a job.
- Select the finished water job in AMSjobsJob → Add to ParAMS (Ctrl-T)This brings up a dialog with a few different options.

- Set Use ResultsImporter to Add Single Job. This will add a single job to the job collection, based on the final structure of the geometry optimization.Set Task (for new job) to GeometryOptimization.Make sure the Properties are emptyClick Import
This adds the job water (or water_1 if you first added the charges and forces - job names need to be unique).
It is marked in red because it is not yet used in any training set entry.
On the Jobs panel, click twice at the job name and change the name to water_geoopt.

Then, add one of the O-H bond lengths:
- Select the
water_geooptjob on the Jobs panel.In the molecule area on the left, select the O atom and one of the H atomsTraining Set → Add → Bonds
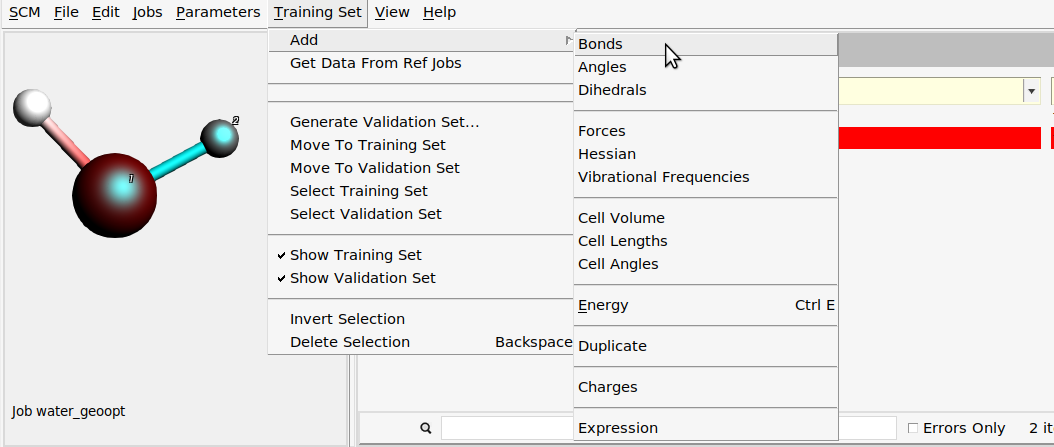
- The job is now no longer marked in redSwitch to the Training Set panel

- An entry of type Geo: distance has been added with the details water_geoopt, 0, 1 (H-O). This means that the bond is between atoms 0 and 1 (in ParAMS, atom indexing starts with 0), which is an O-H bond.The value from the reference job is given in the Value column: 0.99 Å.Select the Geo: distance entry. It will bring up the molecule in the left area with the two atoms selected.
To add the H-O-H angle:
- Select the
water_geooptjob on the Jobs panel.In the molecule area on the left, select one H atom, hold Shift and click on the O atom, hold Shift and click on the final H atomTraining Set → Add → AngleSwitch to the Training Set panel

- An entry of type Geo: angle has been added with the details water_geoopt, 1, 0, 2 (H-O-H).The value from the reference job is given in the Value column: 104.4°.
You can also add dihedral angles in a similar way (for molecules with at least 4 atoms).
2.3.5. PES scans: bond scan, angle scan, or volume scan¶
- Bond scan: See the ReaxFF: Gaseous H₂O tutorial
- Angle scan: in the same way as importing a bond scan
- Dihedral scan: in the same way as importing a bond scan
- Volume scan: in the same way as importing a bond scan. See also the GFN1-xTB: Lithium fluoride tutorial.
For the above type of PESScan reference calculations, there are two options for Use ResultsImporter in the import dialog:
- Add Single Job. This, together with Task (for new job) → PESScan, sets up an equivalent PESScan to be run during the parameter optimization. This is illustrated in the ReaxFF: Gaseous H₂O tutorial.
- Add PESScan Singlepoints. This, together with Task (for new job) → SinglePoint sets up several singlepoint calculations on the frames from the PESScan to be run during the parameter optimization. This is illustrated in the GFN1-xTB: Lithium fluoride tutorial.
| Add Single Job | Add PESScan Singlepoints | |
|---|---|---|
| Task (for new job) | PESScan | SinglePoint |
| Properties | pes | relative_energies, forces, stresstensor |
| Recommended | yes | no (unless necessary) |
| Speed | slow | fast |
| Number of new jobs | 1 | = number of frames |
| Energy curves in GUI | yes | no |
| Tutorial example | ReaxFF: Gaseous H₂O | GFN1-xTB: Lithium fluoride |
The pes property for Add Single Job is equivalent to the
relative_energies property for Add PESScan Singlepoints. They extract
the relative energy of the different frames (by default, relative to the
minimum energy in the scan). It is the fundamental property to extract from a
PES Scan (bond scan, angle scan, volume scan).
With Add PESScan Singlepoints, you can also add the forces and
stresstensor for each frame, but only if you enabled the Save results for
all PES points option when running the PES Scan. See the GFN1-xTB: Lithium fluoride
tutorial for an example.
With Add Single Job (and Task PESScan), many constrained geometry optimizations are performed for each set of parameters, which can be time-consuming. With Add PESScan Singlepoints, only single-point calculations are performed, which is usually faster.
With Add Single Job (and Task PESScan, pes extractor), the GUI will
give useful plots of the energy vs. the scan coordinate for both the reference
values and calculated values during the parametrization. For this reason, we
recommend to use this option and not Add PESScan Singlepoints.
2.3.6. Energies¶
For Lennard-Jones, ReaxFF, and DFTB you typically only want to fit relative energies (reaction energies), not absolute energies. This is because the absolute energy levels are different for different engines.
Important
Note that the absolute energy will be added if you select select the
property energy in the Results Importer dialog, or if you choose
Training Set → Add → Energy. Make sure to edit the entry to become a
relative energy instead!
As an example, let’s add the reaction energy for propene combustion: C₃H₆(g) + (9/2) O₂(g) → 3 CO₂(g) + 3 H₂O(g). First, run the reference calculations and import the jobs:
- Run three more calculations in exactly the same way as for Reference calculation #1: Geometry optimization of a water molecule, but for the molecules propene (C₃H₆), O₂, and CO₂. (And H₂O if you did not run that calculation yet) Save them with the names
propene.ams,o2.ams, andco2.ams, and run them.In AMSjobs, select all three new calculations and choose Job → Add to ParAMS (Ctrl-T).

- In the dialog, set Task (for new job) to Geometry Optimization (depending on your preference).Make sure the Properties list is empty.Click Import All.

- This adds the three new jobs to the Jobs panel in the ParAMS GUI. They are highlighted in red because they are not yet used in any training set entry.
Then, add the reaction energy. You can either work out the stoichoimetric coefficients yourself, or use the automatic Balance feature of ParAMS (see below).
- In the top table, select the
co2job, and choose Training Set → Add → Energy (Ctrl-E).This adds the absolute energy of the CO₂ job. We need to edit it to make into a relative energy (reaction energy).Go to the Training Set panel

- Double-click in the Details column where it says
co2.In the Energy text box, typeenergy("co2")+energy("wand press TAB multiple times. This will cycle through all jobs starting with “w”. If you followed both the Charges and forces and Bonds and angles steps, there will be two jobswaterandwater_geoopt.Continue editing the Energy text box until it readsenergy("co2")+energy("water_geoopt")-1.0*energy("propene")-energy("o2")(start with the products and positive signs, and end with the reactants and negative signs). Write an explicit1.0*beforeenergy("propene")

- Click the Balance button in the bottom left corner. This balances the equation to read
+3.0*energy("co2")+3.0*energy("water")-1.0*energy("propene")-4.5*energy("o2"). Here the coefficient before propene is 1.0 as specified in the previous step.

- Remove the Value. It will then be recalculated from the reference jobs when you click “OK”Click OKOn the Training Set panel, the reaction energy becomes -0.055 eV (it may also be shown in another unit).

Tip
To recalculate reference values after you have modified an expression you can also use the Training Set → Get Data From Ref Jobs option.
This is the reaction energy calculated at the UFF (ForceField) level of theory - it is very far from reality! For a more realistic reference value, run the reference calculations with DFT (ADF, BAND, or Quantum ESPRESSO) instead.
For PES Scans, the pes and relative_energies properties extract relative energies from the PES scan.
See also
The Import experimental formation enthalpy into ParAMS of the GFN1-xTB tutorial explains how to import experimental reaction energies (i.e., not calculated from reference jobs), and also has some tips for reaction energies in general.
2.3.7. Import a molecular dynamics trajectory¶
You can import frames from a Molecular Dynamics trajectory. Typically, running a molecular dynamics is a simple way of sampling some distorted structures, and can be used for fitting especially the forces but also relative energies.
There are two ways:
- Run the MD simulation directly with the reference method (often DFT, i.e. you would run ab initio MD).
- Run the MD simulation with a parametrized method, use the Replay task of the AMS Driver to recalculate some of the frames with DFT, and add only the DFT-calculated frames to ParAMS. In particular, because training sets often need to be expanded iteratively, the initial MD simulation could be run with your current force field, if you have already run a parametrization.
2.3.7.1. Reference calculation #2: MD simulation of liquid Ar¶
- Open AMSinput, switch to the panel.File → Import coordinates, select the file
Ar32.xyzSet Task to Molecular DynamicsClick the next to the TaskSet Number of steps to
next to the TaskSet Number of steps to70.Set Timestep to5fs.Set Sample frequency to10.Set Initial temperature to2000K.


- Click the next to Trajectory OptionsSet Write engine gradients to Yes. The “engine gradients” are the negative of the atomic forces. If you do not check this option, the gradients (negative forces) will not be saved to the trajectory and cannot be imported to ParAMS.

- Save with the job
uff_mdRun the calculation. It should finish in a few seconds.If a dialog asks you to update the coordinates, select No.
You can select the job in AMSjobs and select SCM → Movie to visualize the trajectory.
Note
The initial velocities are random. The trajectory (and the forces on the atoms) will be different when you run the calculation yourself compared to the screenshots in this tutorial.
2.3.7.2. MD with reference method¶
Here, we will use the previous calculation as an MD simulation with the
reference method. Normally, you would have instead used DFT (on the  or
or  panels, or using Quantum ESPRESSO or VASP) for the MD simulation.
panels, or using Quantum ESPRESSO or VASP) for the MD simulation.
- Select the finished
uff_mdjob in AMSjobs.Job → Add to ParAMS (Ctrl-T)In the dialog, select Use ResultsImporter: Add Trajectory SinglePoints.Set Steps from 3 to 7Set Step size to 2. This will import frames 3, 5, and 7 from the trajectory.Set Properties to relative_energies and forces. Note: you can select multiple entries in the drop-down list!
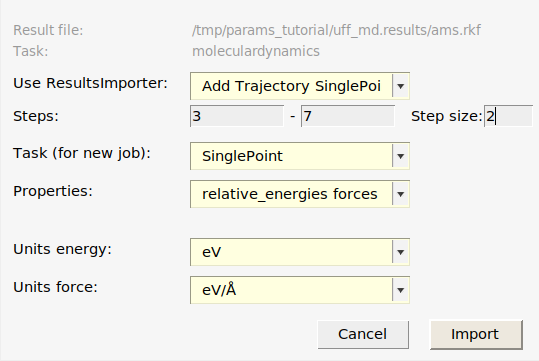
- Click Import

This adds:
- Three Singlepoint jobs to the job collection:
uff_md_frame003,uff_md_frame005, anduff_md_frame007, containing the structures of those frames. - Two relative energy expressions:
uff_md_frame003-uff_md_frame005anduff_md_frame007-uff_md_frame005, with the corresponding reference values. In this case, frame 5 had the lowest energy so the relative energies are taken relative to that frame. In your case, frame 3 or 7 may be the lowest energy frame. - Three forces expressions for the three frames. If you select a force entry and switch to the Info panel in the bottom table, you can see the individual force components.
- An Engine containing the settings for the reference engine (in this case UFF, which corresponds to an empty [default]
Engine ForceFieldblock).
2.3.7.3. MD with fast method followed by Replay with reference method¶
The task Replay performs a series of single-point calculcations on structures taken from an ams.rkf file, for example a molecular dynamics trajectory.
Here, we will use the GFN1-xTB (DFTB) method to recalculate some of the points from the molecular dynamics trajectory generated with UFF. If you do not have a DFTB license, you could instead run the Replay calculation with BAND, or even ForceField (UFF).
- SCM → New InputSwitch to the
 panel (or use a different engine if you do not have a DFTB license)Task → ReplayClick the arrow next to the TaskClick the icon next to Restart from and browse to the
panel (or use a different engine if you do not have a DFTB license)Task → ReplayClick the arrow next to the TaskClick the icon next to Restart from and browse to theams.rkffile from the previous job. It is located inside the folderuff_md.results.Set Frames to3 5 7

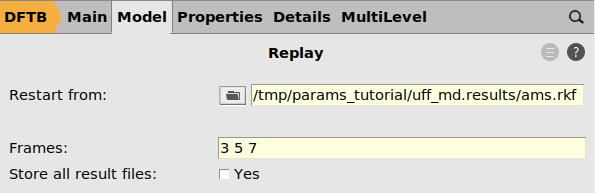
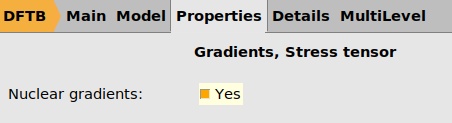
- Go to the Properties → Gradients, Stress tensor panel.Set Nulear gradients to Yes. This is needed to save the calculated forces so that they can be imported into ParAMS.
- Save the job with the name
Ar32.amsand run the job.Wait for the job to finish.If you get a dialog asking you to completely replace the system, select No.
When the calculation has finished, import it into ParAMS:
- Select the job in AMSjobsJob → Add to ParAMS (Ctrl-T)In the dialog, select Use ResultsImporter: Add Trajectory SinglePoints.Set Steps from 1 to 3Set Step size to 1. This will import frames 1, 2, and 3 from the Replay job (corresponding to frames 3, 5, and 7 from the original trajectory).Set Properties to relative_energies and forces. Note: you can select multiple entries in the drop-down list!

- Click Import
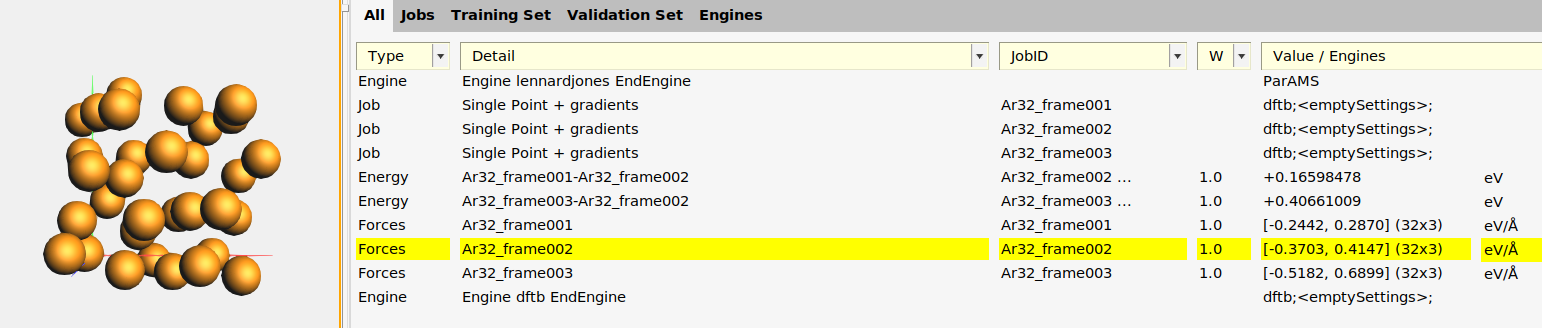
This adds:
- Three Singlepoint jobs to the job collection:
Ar32_frame001,Ar32_frame002, andAr32_frame003, containing the structures of those frames. - Two relative energy expressions:
Ar32_frame001-Ar32_frame002andAr32_frame003-Ar32_frame002, with the corresponding reference values. In this case, frame 2 had the lowest energy so the relative energies are taken relative to that frame. In your case, frame 1 or 3 may be the lowest energy frame. - Three forces expressions for the three frames. If you select a force entry and switch to the Info panel in the bottom table, you can see the individual force components.
- An Engine containing the settings for the reference engine (in this case GFN1-xTB, which corresponds to an empty [default]
Engine DFTBblock).
The above is exactly how the training data for the Getting Started: Lennard-Jones Potential for Argon tutorial was created.
2.3.8. Import a structure and settings from AMSinput¶
You can import a structure and AMS driver settings (for example, geometry optimization settings) from AMSinput. This can be useful if you
- want to import an experimental structure
- want to import a structure optimized with a DFT code not supported by ParAMS (codes other than AMS, VASP, or Quantum ESPRESSO)
See the Import experimental formation enthalpy into ParAMS section of the GFN1-xTB tutorial for an example of how to do this.
Note
You often want to set Task GeometryOptimization when importing from AMSinput. You should then also set the Maximum number of iterations on the Details → Geometry Optimization panel and the Pretend converged option on the Expert AMS panel. See the Frequently Asked Questions for details.
2.3.9. Import VASP or Quantum ESPRESSO calculations¶
You can import results from VASP or Quantum ESPRESSO single points, geometry optimizations, and molecular dynamics simulations.
Note
If you ran VASP via AMS (the  in AMSinput), there should be an ams.rkf file in the results directory. In that case, follow the previous instructions on this page.
in AMSinput), there should be an ams.rkf file in the results directory. In that case, follow the previous instructions on this page.
For VASP calculations run outside AMS, open the ParAMS GUI and select Jobs →
Add Job, select the OUTCAR file from the file dialog. This will convert the
VASP output to AMS format - it may take a while. Then, you will see the
familiar results importer dialog, where you can choose either Add Single
Job or Add Trajectory Singlepoints, and select properties including
energy, forces, and stresstensor.
For Quantum ESPRESSO calculations, open the ParAMS GUI and select Jobs → Add
Job, select the jobname.out file from the file dialog. This will convert
the output to AMS format - it may take a while. Then, you will see the
familiar results importer dialog, where you can choose either Add Single
Job or Add Trajectory Singlepoints, and select properties including
energy, forces, and stresstensor.
Note
If you run Quantum ESPRESSO outside the AMS GUI, it is necessary to
redirect the standard output to a file with a name ending in .out
when you run the calculation.
For example:
/path/to/pw.x < jobname.in > jobname.out
See also
The page AMS, VASP and Quantum ESPRESSO reference data explains where the results from the conversion to AMS format are stored, and the meaning of the added engines to the engine collection. In particular, the added engines cannot be used for new reference calculcations.