Generating multiple conformers for a compound¶
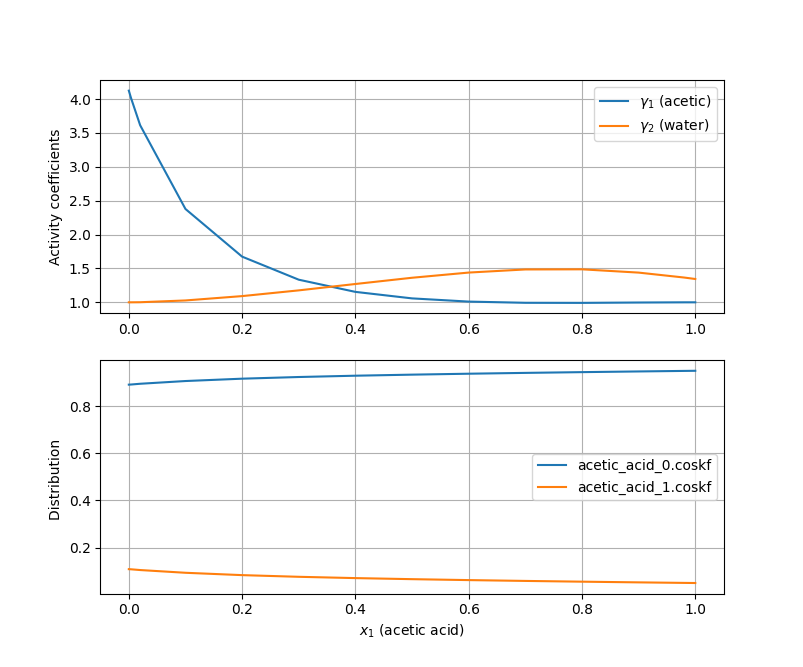
Different conformers of a molecule can have significantly different sigma profiles, which can lead to big differences in predicted properties with COSMO-RS. For this reason, it’s important for COSMO-RS calculations to use geometries corresponding to the lowest-energy conformer or a set of low-energy conformers when it’s possible several conformers may exist in significant amounts. The script below allows the user to use the ConformerTools functionality in AMS to generate a set of low-energy conformers, refine the geometries with a semi-empirical method, and finally perform the ADF and COSMO calculations necessary to produce .coskf files. The example files are used in a simple binary mixture calculation with COSMO-RS. This calculation models one of the species as a mixture of conformers and plots activity coefficients and the conformer distribution over the mole fraction range.
Python code (Binary mixture)¶
[show/hide code]
import os
import matplotlib.pyplot as plt
from scm.conformers import ConformersJob
from scm.plams import from_smiles, init, finish, Settings, delete_job, KFFile, AMSJob, Molecule, CRSJob, Units
class COSKFConformers:
'''
Args:
mol_identifier (str) : A smiles string or file (.xyz, .pdb, .mol, or mol2) with which to input a molecule
Keyword Args:
generator (str) : The method used to generate conformers. Can be one of ['RDKit','CREST']
initial_conformers (int) : The number of initial conformers to sample
coskf_name (str) : the filename base for the .coskf files this script produces. multiple conformers will be saved with the filename base appended with a ``_n`` where n is the index of the conformer.
max_energy_range (float) : the largest acceptable energy (relative to the lowest energy conformer) for an adf calculation to be performed. The energy is compared at the dftb level.
max_num_confs (int) : the maximum number of conformers to produce .coskf files for
print_output (bool) : Whether to print more detailed information about the progress of the script.
dftb_solvent (str) : A solvent to use with GFN-xTB (DFTB). Can be one of ['Acetone','Acetonitrile','CHCl3','CS2','DMSO','Ether','H2O','Methanol','THF','Toluene']
coskf_dir (str) : The directory where produced .coskf files should be saved. Defaults to the <plams run directory>/conformers
'''
def __init__(self,
mol_identifier : str,
generator = 'RDKit',
initial_conformers = 500,
coskf_name = None,
max_energy_range = 2.0,
max_num_confs = None,
print_output = False,
dftb_solvent = None,
coskf_dir = None
):
self.print_output = print_output
self.coskf_name = coskf_name
self.generator = generator
self.initial_conformers = initial_conformers
self.dftb_solvent = dftb_solvent
self.coskf_map = {}
self.coskf_dir = coskf_dir
self.mol = self.process_molecule(mol_identifier)
if self.mol is None:
print("There was a problem reading the molecule: ", mol_identifier)
return
self.confs0 = self.get_conformers()
self.confs1 = self.conformers_xtb()
if max_num_confs and len(self.confs1.get_relative_energies())>max_num_confs:
num_max_energy = self.confs1.get_relative_energies()[max_num_confs-1]
num_max_energy = Units.convert(num_max_energy,'Hartree','kcal/mol') + 0.001
max_energy_range = min(max_energy_range,num_max_energy) if max_energy_range else num_max_energy
self.confs2 = self.conformers_adf(max_energy_range=max_energy_range)
replay = self.conformers_cosmo()
self.make_coskfs(replay)
# Remove the results from the replay job because they can be large
delete_job(replay.job)
def process_molecule(self, mol_identifier):
'''
This function tries to process any molecular identifier passed to the COSKFConformers constructor.
This first checks for a filename ending with .xyz, .pdb, .mol, or mol2. If this is not found, the
molecule is assumed to be input as a SMILES string.
'''
if any( mol_identifier.endswith(x) for x in ['.xyz','.pdb','.mol','mol2'] ):
try:
return Molecule(mol_identifier)
except:
return None
try:
return from_smiles(mol_identifier)
except:
return None
def get_conformers(self):
'''
Optimize conformers at the UFF level
'''
sett = Settings()
if self.generator == 'RDKit':
sett.input.AMS.Generator.RDKit
sett.input.AMS.Generator.RDKit.InitialNConformers = self.initial_conformers
conformers_uff = ConformersJob(name="conformers_uff", molecule=self.mol).run()
if self.print_output:
print("Conformers at the UFF level:")
print(conformers_uff)
return conformers_uff
def conformers_xtb(self):
'''
Optimize conformers with DFTB
'''
sett = Settings()
sett.input.AMS.Task = "Optimize"
sett.input.AMS.InputConformersSet = self.confs0
sett.input.DFTB
if self.dftb_solvent:
sett.input.DFTB.Solvation.Solvent = self.dftb_solvent
conformers_xtb = ConformersJob(name="conformers_xtb", settings=sett).run()
if self.print_output:
print(f"Conformers at the GFN1-xTB level with solvent {self.dftb_solvent}:")
print(conformers_xtb)
return conformers_xtb
def conformers_adf(self,max_energy_range=None):
'''
Optimize conformers with ADF and the special settings for CRS
'''
sett = Settings()
sett.input.AMS.Task = "Optimize"
sett.input.AMS.GeometryOptimization.UseAMSWorker = "False"
sett.input.AMS.InputConformersSet = self.confs1
self._add_default_crs_settings(sett)
if max_energy_range:
sett.input.AMS.InputMaxEnergy = f'{max_energy_range} [kcal/mol]'
conformers_adf = ConformersJob(name="conformers", settings=sett).run()
if self.print_output:
print("Conformers at the DFT level:")
print(conformers_adf)
return conformers_adf
def conformers_cosmo(self):
'''
Replay job on the conformer set to generate all the ADF engine output files with the COSMO sections.
These are then converted to .coskf files afterwards.
'''
sett = Settings()
self._add_default_crs_settings(sett)
sett.input.AMS.Task = "Replay"
sett.input.AMS.Replay.File = self.confs2["conformers.rkf"]
sett.input.AMS.Replay.StoreAllResultFiles = "True"
sett.input.ADF.Symmetry = "NOSYM"
self._add_default_crs_solvation(sett)
replay = AMSJob(name="replay", settings=sett).run()
return replay
def make_coskfs(self,replay):
'''
Copy the COSMO sections from all the files back to the folder with the conformers
'''
base_name = self.coskf_name if self.coskf_name is not None else "conformer"
if self.coskf_dir is None:
self.coskf_dir = self.confs2.job.path
for i, E in enumerate(self.confs2.get_energies("Ha")):
if f"Frame{i+1}.rkf" in replay:
cosmo_section = replay.read_rkf_section("COSMO", f"Frame{i+1}")
cosmo_section["Gas Phase Bond Energy"] = E
name = f"{base_name}_{i}.coskf"
coskf = KFFile(os.path.join(self.coskf_dir, name), autosave=False)
for key, val in cosmo_section.items():
coskf.write("COSMO", key, val)
coskf.save()
self.coskf_map[i] = name
def _add_default_crs_settings(self,sett):
'''
This function adds the default settings for optimizing geometries in cosmo-rs/-sac.
'''
sett.input.ADF.Basis.Type = "TZP"
sett.input.ADF.Basis.Core = "Small"
sett.input.ADF.BeckeGrid.Quality = "Good"
sett.input.ADF.XC.GGA = "BP86"
def _add_default_crs_solvation(self,sett):
'''
This function adds the solvation block required for generating sigma profiles.
Note that in this section, only the radii of the most common elements have been added. If your compound
contains an element not listed below, you'll have to add its symbol and radius in the same format as the others.
'''
sett.input.ADF.Solvation = {
"Surf": "Delley",
"Solv": "Name=CRS Cav0=0.0 Cav1=0.0",
"Charged": "Method=Conj Corr",
"C-Mat": "Exact",
"SCF": "Var All",
"Radii": {
"H": 1.30,
"C": 2.00,
"N": 1.83,
"O": 1.72,
"F": 1.72,
"Si": 2.48,
"P": 2.13,
"S": 2.16,
"Cl": 2.05,
"Br": 2.16,
"I": 2.32,
},
}
init()
coskf_name = "acetic_acid"
conformers = COSKFConformers(
'CC(=O)O',
print_output=True,
coskf_name = coskf_name,
max_energy_range=None,
max_num_confs=100,
dftb_solvent=None,
coskf_dir='conformer_coskfs')
# here we'll do a simple cosmo-rs calculation with the conformers produced above
# this will be a binary mixture calculation with water (available in the cosmo-rs compound database)
settings = Settings()
settings.input.property._h = 'BINMIXCOEF'
# absolute paths are usually easier for input
path_to_confs = os.path.abspath(conformers.coskf_dir)
num_compounds = 2
compounds = [Settings() for i in range(num_compounds)]
compounds[0].name = "acetic_acid"
form = [Settings() for i in range(len(conformers.coskf_map))]
for i,v in conformers.coskf_map.items():
form[i]._h = os.path.join( path_to_confs, v )
compounds[0].form = form
compounds[1].name = "water"
compounds[1]._h = os.path.join( os.getcwd(), "Water.coskf" )
settings.input.temperature = 298.15
settings.input.compound = compounds
crs_job = CRSJob(settings=settings)
out = crs_job.run()
res = out.get_results()
comp_acetic = out.get_multispecies_dist()[0]
mf1 = res['molar fraction'][0]
fig, axs = plt.subplots(2)
axs[0].plot( mf1, res['gamma'][0], label = '$\gamma_1$ (acetic)')
axs[0].plot( mf1, res['gamma'][1], label = '$\gamma_2$ (water)')
for struct, val in comp_acetic.items():
axs[1].plot( mf1, val, label = os.path.basename(struct))
plt.setp(axs[0],ylabel='Activity coefficients')
plt.setp(axs[1],ylabel='Distribution ')
axs[0].legend()
axs[0].grid()
axs[1].legend()
axs[1].grid()
plt.xlabel('$x_1$ (acetic acid)')
plt.show()
finish()
This code produces the following figure which plots activity coefficients and the distribution of two conformers.