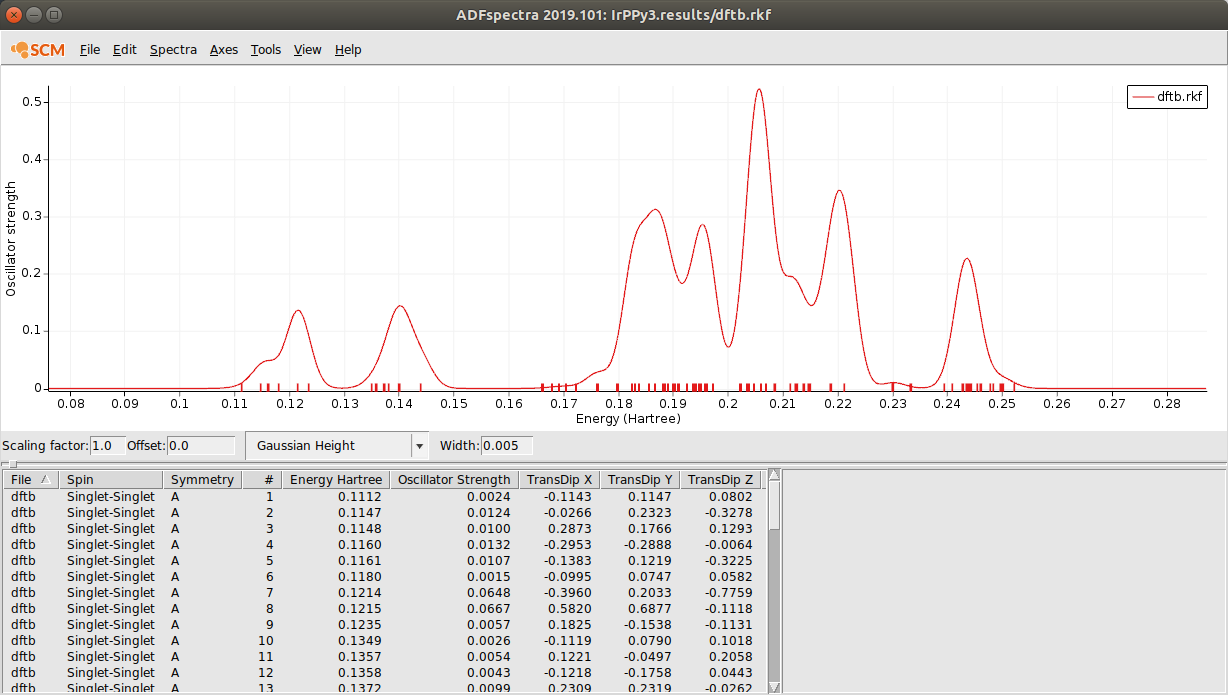
UV/Vis spectrum of Ir(ppy)3¶
In this tutorial we will use time-dependent DFTB to calculate the UV/Vis absorption spectrum of the Iridium complex Ir(ppy)3.
- Start ADFinput.Obtain a Ir(ppy)3 molecule by copy-pasting the following coordinates into ADFinput:
Ir 0.012398 0.011138 -0.034888
N -0.039454 0.033020 2.182572
C 1.196760 -0.025409 2.759734
C 1.312058 0.049492 4.160403
H 2.297136 0.010793 4.623158
C 0.176907 0.180049 4.949521
H 0.267945 0.241793 6.035562
C -1.080728 0.227837 4.337521
H -1.998559 0.321169 4.918229
C -1.136029 0.147902 2.951293
H -2.086922 0.175812 2.417126
C 2.320171 -0.152466 1.829294
C 2.023265 -0.155989 0.436568
C 3.102011 -0.288673 -0.454369
H 2.909074 -0.300226 -1.528367
C 4.415258 -0.411210 0.002305
C 4.695599 -0.406624 1.375984
H 5.722404 -0.504771 1.732389
C 3.649435 -0.277487 2.282572
H 3.871783 -0.280893 3.351436
H 5.231463 -0.511562 -0.717544
C 0.348035 0.181591 -2.073796
C 0.477555 1.514919 -2.556751
C 0.724684 1.764358 -3.922454
H 0.829885 2.787826 -4.287727
C 0.844594 0.712499 -4.823508
H 1.037805 0.912270 -5.878913
C 0.719659 -0.604432 -4.359020
H 0.813485 -1.438052 -5.059452
C 0.474019 -0.859703 -3.009006
H 0.385255 -1.893716 -2.671738
C 0.365837 2.600714 -1.582092
N 0.167789 2.211524 -0.287922
C 0.070484 3.136313 0.682683
H -0.072051 2.747901 1.692346
C 0.145763 4.501897 0.436809
C 0.336880 4.923011 -0.884173
H 0.401581 5.985888 -1.124354
C 0.445887 3.971689 -1.889620
H 0.597414 4.283773 -2.922014
H 0.063205 5.212485 1.259290
N -2.184281 -0.109350 -0.312568
C -2.661123 -1.388612 -0.350406
C -4.046619 -1.609170 -0.458307
H -4.429551 -2.628420 -0.482142
C -4.923390 -0.534729 -0.529917
H -5.997987 -0.707636 -0.612774
C -4.412321 0.767839 -0.500666
H -5.061792 1.641013 -0.565173
C -3.036095 0.927439 -0.392594
H -2.577985 1.917423 -0.370095
C -1.646438 -2.441110 -0.271132
C -0.290616 -2.036409 -0.112573
C 0.684523 -3.046718 -0.059045
H 1.733837 -2.770376 0.056220
C 0.343229 -4.396378 -0.157833
C -0.995273 -4.783541 -0.314549
H -1.261180 -5.838892 -0.394860
C -1.983096 -3.806784 -0.368630
H -3.023159 -4.112311 -0.498560
H 1.126462 -5.157297 -0.113713
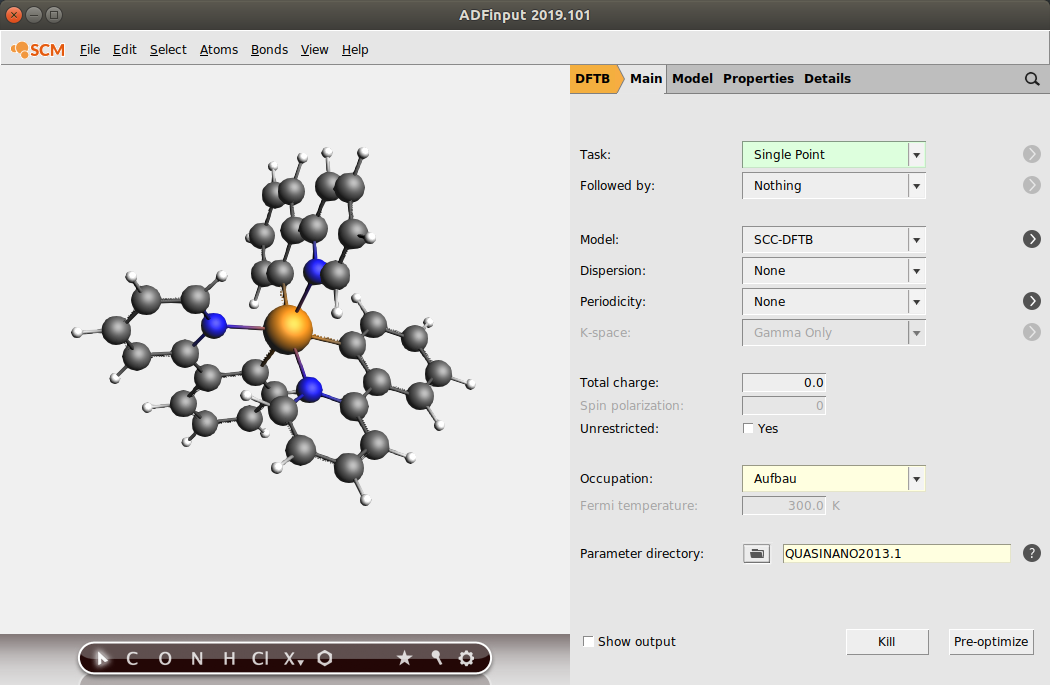
- Select the DFTB panel: Panel bar ADF → DFTB.Choose Single Point as the task to perform.
TD-DFTB is based on the SCC extension to DFTB and is therefore best used with the SCC-DFTB model. It is also incompatible with fractional occupation numbers, so we switch to the Aufbau occupation scheme.
- Make sure the model is set to SCC-DFTB.Select the Aufbau occupation scheme.
We need to select a parameter set that includes Iridium.
- Change the parameter directory to QUASINANO2013.1
Your ADFInput window should look like this:
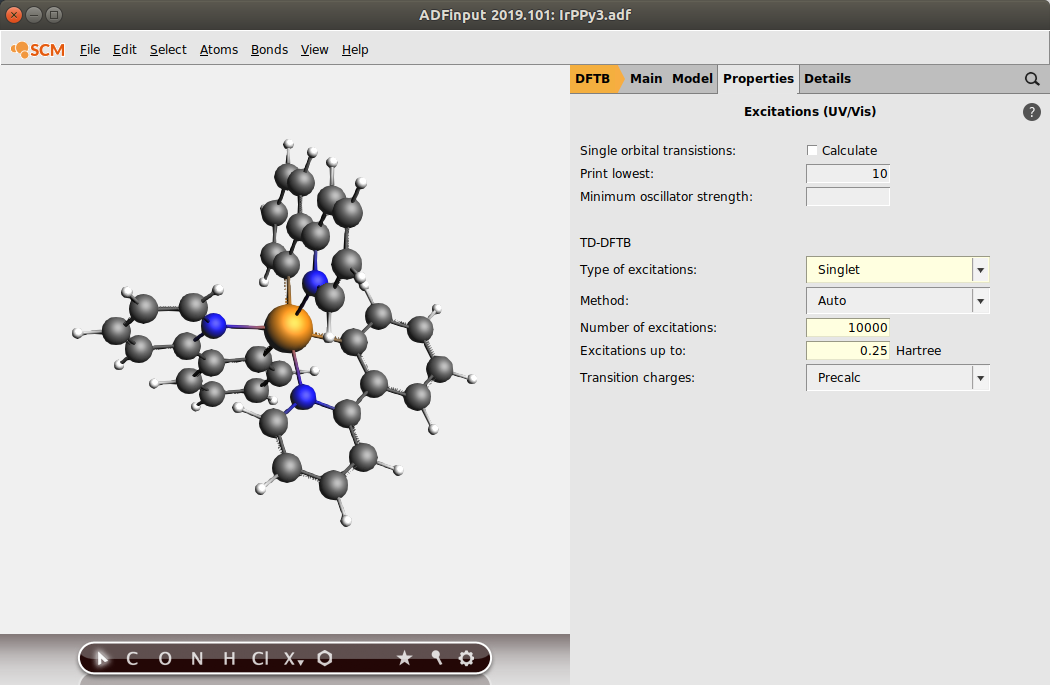
We want to obtain the absorption spectrum up to 6 eV, so we allow for some safety margin and calculate all singlet-singlet excitations up to 6.8 eV which is 0.25 Hartree. The “Number of excitations” field acts an upper limit for the calculated number of excitations if the “Excitations up to” field is used. We do not need an upper limit for this tutorial, so we set it to something large.
- Use the panel bar Properties → Excitations (UV/Vis) to go to the TD-DFTB configuration.TickSelect Singlet as the Type of excitations to calculate.Change the Number of excitations to calculate to 10000.Calculate Excitations up to 0.25 Hartree.
We are now ready to run the calculation. It should only take a few seconds.
- Save your input using File → Save As....Run the calculation with File → Run.Wait for the calculation to finish.
We can now use ADFspectra to have a look at absorption spectrum.
- Select SCM → Spectra