PES Exploration: Water dissociation on an oxide surface¶
This tutorial will teach you how to
Use basin hopping and process search for molecules adsorbed on surfaces
Find dissociation barriers for water on an oxide surface
See also
A more comprehensive introduction to PES exploration can be found in the Automated reaction pathway discovery for hydrohalogenation tutorial and the PES Exploration documentation.
Introduction¶
Water adsorption at metal oxide surfaces is common in heterogeneous catalysis, electrochemistry, and geochemistry. Water can either adsorb molecularly or dissociate at the surface, and can donate hydrogen bonds to surface atoms and/or other water molecules.
One well-studied system is water adsorption on ZnO(\(10\bar{1}0\)). A monolayer of H2O can adsorb molecularly, dissociatively, or in a 50/50 (half-dissociated) configuration.
Raymand et al. developed a ReaxFF force field for H2O adsorption on ZnO. Here, we will use this force field to illustrate how basin hopping and process search can be used to automatically discover the three most stable types of water monolayer adsorption, and the barriers for converting from one structure to the other.
Outline:
Step 1: import a model of two water molecules adsorbed on ZnO(\(10\bar{1}0\)).
Step 2: Run basin hopping (a type of global optimization) to quickly discover several local minima, including the half-dissociated configuration
Step 3: Run process search on the half-dissociated configuration to find the barriers for converting to the fully molecular or fully dissociated configurations.
Step 1: Set up the initial system¶
Import the system into AMSinput¶
Download 2H2O_ZnO_10-10_2dl.xyz
and import it into AMSinput. The 2D slab was constructed from the
ReaxFF-optimized lattice parameters a = 3.28 Å and c = 5.28 Å (see Raymand
et al.). It is a (2×1) supercell of the ZnO(\(10\bar{1}0\)) surface unit cell, with x
|| [\(1\bar{2}10\)] and y || [0001]. The slab is 2 double-layers thick.
The two H2O molecules were manually added to reasonable adsorption positions. This is only a starting point for the global optimization, so it is not too critical exactly how the water molecules are positioned.
See also
More information about creating slabs and orienting molecules on surfaces can be found in the Crystals and Surfaces tutorial.
Note
To find the half-dissociated configuration, at least two water molecules must be modeled. This is why a (2×1) surface supercell is used.
Change the default color of Zn atoms¶
In AMSinput, the default colors of both Zn and H are white. This can make it difficult to distinguish the atoms. Therefore, we will change the color of Zn to brown.
 plus sign next to Default Atom Colors.
plus sign next to Default Atom Colors.#de6b00 into the Selection: text box and press Enter, or use the sliders to choose your own color.Step 2: Basin Hopping¶
PES exploration tasks like basin hopping and process search benefit greatly from a smooth potential energy surface. ReaxFF contains several options to smoothen the potential energy surface. Here, we will use the option to taper bond orders.
ReaxFF settings¶
 panel
panelBasin hopping settings¶
 arrow or go to the Model → PES Exploration panel.
arrow or go to the Model → PES Exploration panel.8.8.Keep the bottom side of the slab fixed¶
Keeping the bottom side of the slab (the side without water molecules) fixed prevents the crystal from becoming completely distorted during the global optimization.
to add a new region. This will display a shadow around the selected atoms.Region_1 to bottom_side.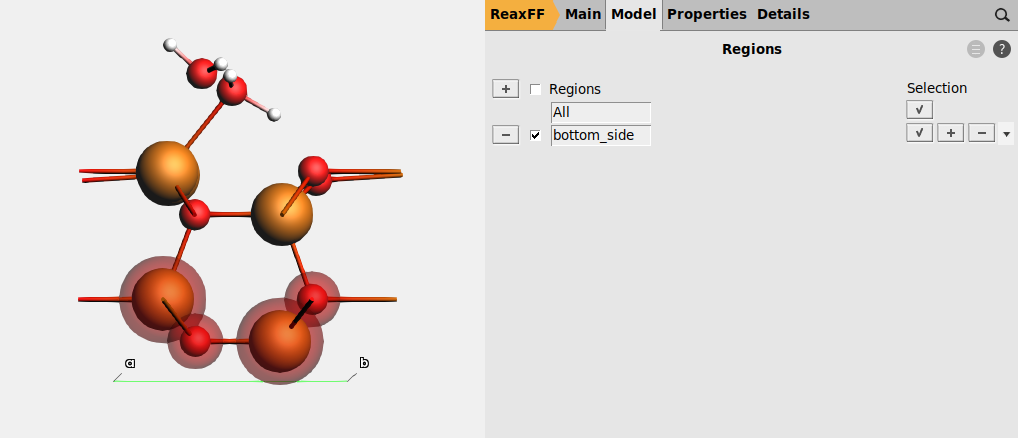 plus sign next to bottom_side (fixed position).
plus sign next to bottom_side (fixed position).
Save and run the basin hopping job¶
basinhopping.amsWait a few minutes for the job to finish.
View the basin hopping results¶
Note
You may not get exactly the same results as are shown in this tutorial. This is because the PES exploration task relies heavily on random numbers.
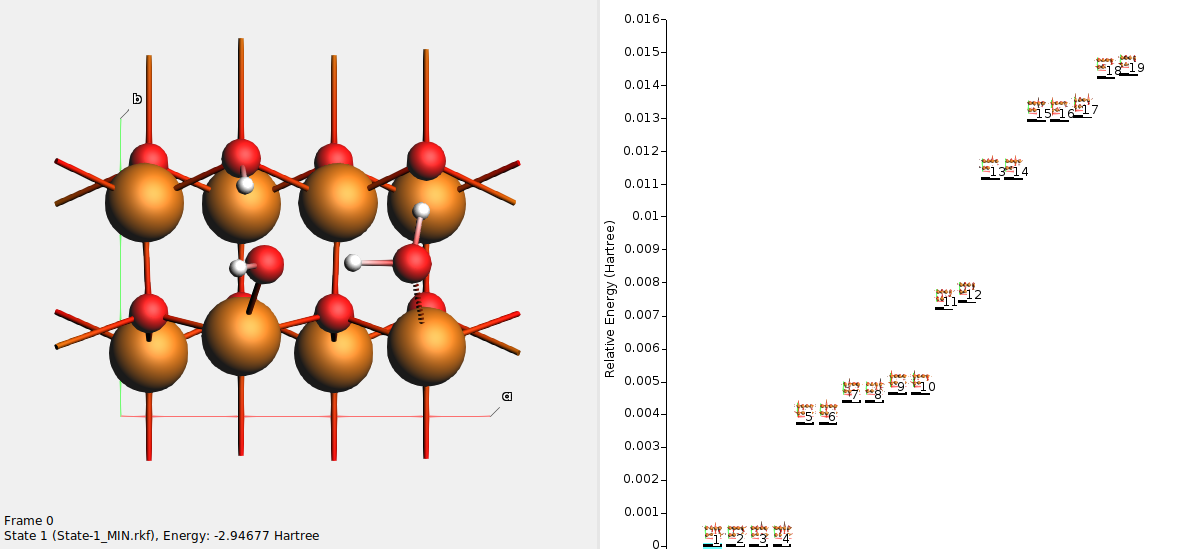
On the right-hand side you see all the states (local minima) found during the basin hopping procedure. In this example, 19 different local minima were found.
Use either the scrollbar at the left, or click on the state lines on the right, to browse the states. Each state has a unique number. The state numbers are sorted with respect to energy, such that state 1 is the lowest-energy state.
Tip
If the structures from different states overlap, use the Energy Profile → Increase Spacing command.
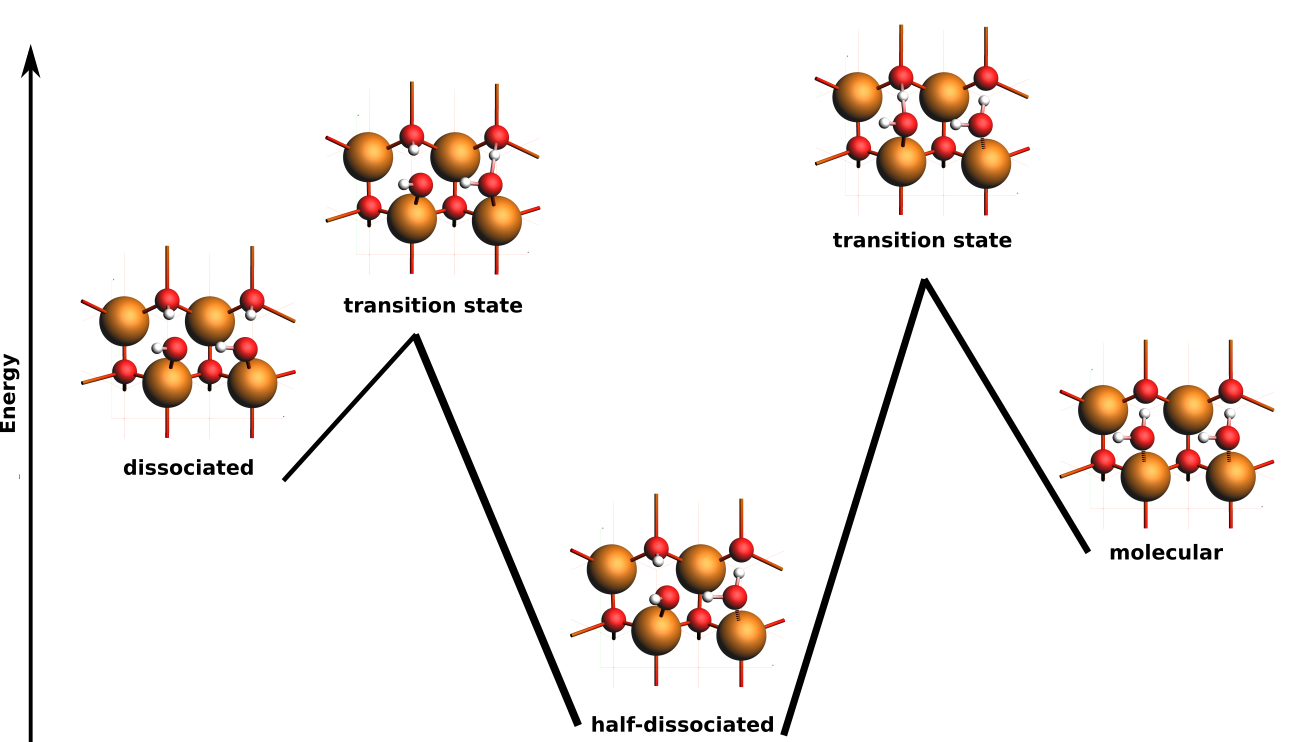
State 1 should correspond to a half-dissociated structure. One of the water molecules has dissociated into OH– adsorbed on a surface Zn, and H+ adsorbed on surface O. The other water molecule donates a hydrogen bond to the OH– and to a surface O.
Here, states 1–4 all correspond to the half-dissociated state. They have the same energy, but differ in which water molecule is dissociated, or in which direction the OH bonds point.
States 5–6 correspond to fully molecular adsorption. The states differ in which direction the OH bonds point.
States 7–8 correspond to a less stable half-dissociated adsorption. The states differ in which direction the OH bonds point.
States 9–10 correspond to fully dissociated adsorption. The states differ in which direction the OH bonds point.
States 11-19 correspond to higher energy (less stable) structures.
Step 3: Process search for reaction barriers¶
For the process search, we will continue from one of the half-dissociated states (1-4) from the previous step.
Tip
Use a state where no water molecule crosses a periodic boundary. This makes visualization easier. In the above example, states 1 and 4 would be suitable.
Process search finds transition states connecting a seed state to nearby local minima.
Here, we are only interested in transition states and minima close to the half-dissociated state. One way to accomplish this is to use the half-dissociated state as the initial state and to only run a single expedition.
Tip
For more advanced ways of continuing from a subset of states from previous calculations, see the Automated reaction pathway discovery for hydrohalogenation tutorial.
process_search.ams116.When the calculation has finished, open the results in AMSmovie: SCM → Movie.
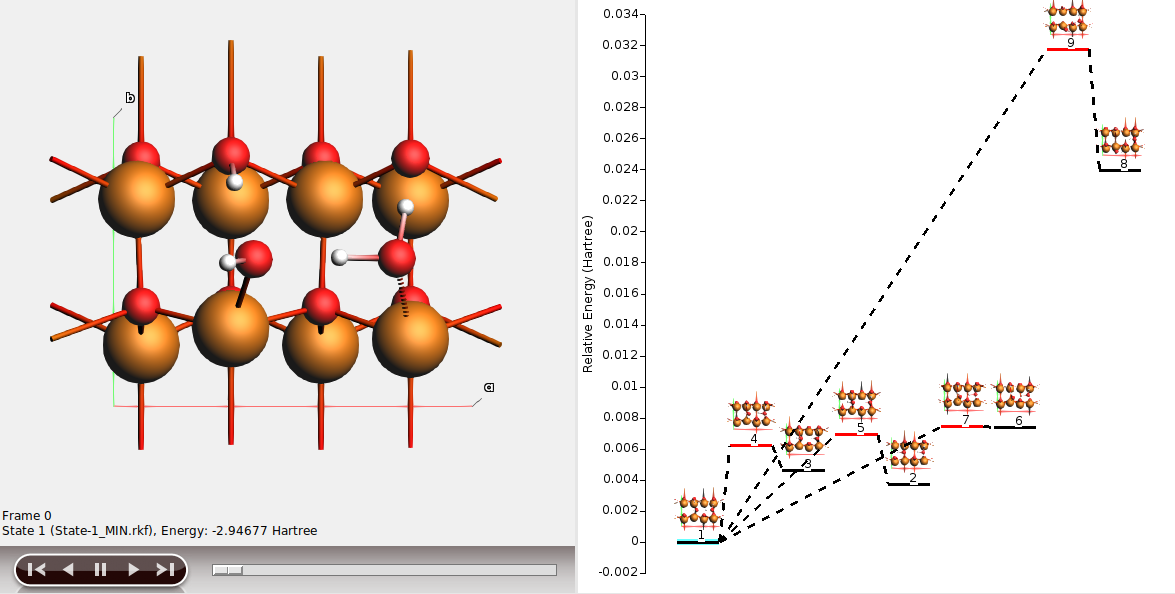
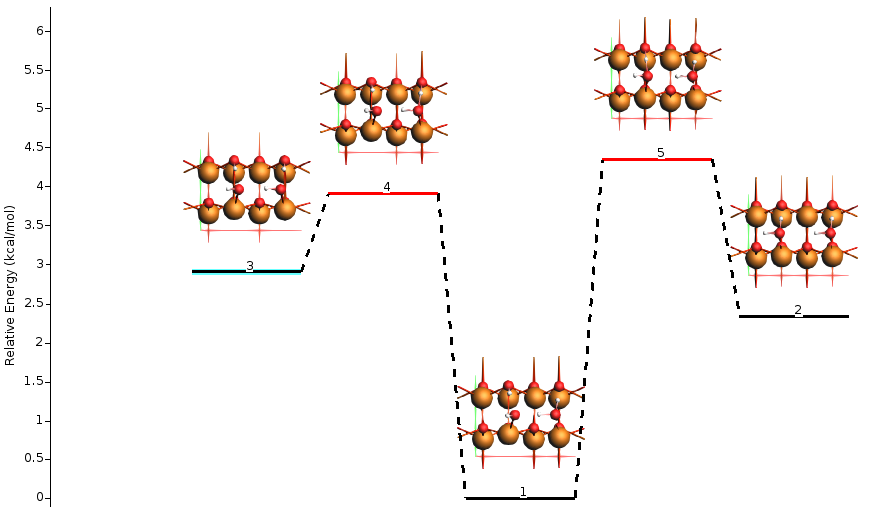
In this example,
state 1 is the half-dissociated state (local minimum)
state 2 is the molecular state (local minimum)
state 3 is the dissociated state (local minimum)
state 4 is the transition state between states 1 and 3
state 5 is the transition state between states 1 and 2
states 6-9 are less stable structures (minima and transition states).
The energy landscape can be rearranged to better highlight the interesting states 1-5.